Myelodysplastic syndrome (MDS) disorders are clonal diseases that often carry stereotypic chromosomal abnormalities. A smaller proportion of cases harbor point mutations that activate oncogenes or inactivate tumor suppressor genes. New technologies have accelerated the pace of discovery and are responsible for the identification of novel genetic mutations associated with MDS and other myeloid neoplasms. These discoveries have identified novel mechanisms in the pathogenesis of MDS. This article touches on the better known genetic abnormalities in MDS and explains in greater detail those that have been discovered more recently. Understanding how mutations lead to MDS and how they might cooperate with each other has become more complicated as the number of MDS-associated genetic abnormalities has grown. In some cases, these mutations have prognostic significance that could improve upon the various prognostic scoring systems in common clinical use.
Myelodysplastic syndromes (MDS) represent a broad group of hematologic disorders characterized by ineffective hematopoiesis, cytopenias, and in many cases, progression to acute myeloid leukemia (AML). Recurrent somatic genomic abnormalities include gains or losses of chromosomal segments, balanced translocations, point mutations, and epigenetic modifications. These lesions alter the differentiation, self-renewal, or proliferation of an MDS stem cell and its progeny. A complete description of the molecular lesions in MDS will enable more accurate prognostic predictions, a molecular taxonomy of MDS, and the identification of novel therapeutic targets in MDS.
Approximately 50% of patients with MDS have diseased cells with an abnormal karyotype. The most common of these cytogenetic alterations are loss of the long arm of chromosome 5 (-5q), loss of chromosome 7 or 7q (-7/7q-), deletion of chromosome arm 20q (20q-), and gain of chromosome 8 (+8) ( Fig. 1 ). The International Prognostic Scoring System (IPSS) for MDS incorporates the most common karyotypic abnormalities into its risk score, attributing the worst risk to patients that carry three or more chromosomal irregularities. Less is known about how these large regions contribute to disease pathogenesis at a molecular level, but some insights have been made recently.
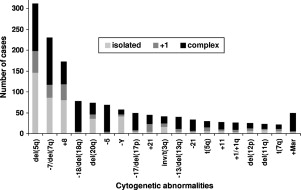
The analysis of mononuclear cell karyotypes is currently the only genetic test in routine clinical use for MDS diagnosis. However, many cases harbor submicroscopic deletions or amplifications, acquired uniparental disomy, and point mutations that can have prognostic significance. The IPSS, although slightly outdated, is the most commonly used prognostic index clinically. The IPSS includes a subset of common chromosomal abnormalities, but does not take into account these more cryptic mutations. For example, point mutations causing the activation of RAS genes or the inactivation of RUNX1 or P53 are well described in MDS cases with normal and abnormal cytogenetics. As the list of genes found to be recurrently mutated in MDS has grown, the biologic and clinical consequences of these mutations has not been fully elucidated. Thus, they are not yet considered in clinical practice. Similarly, the role of epigenetic modifications in the pathogenesis of MDS has been highlighted by the clinical efficacy of hypomethylating agents. However, molecular predictors of response are not yet established. With the genomic analysis of larger patient cohorts, stronger clinical correlations may be possible and more detailed molecular analysis of patients with MDS may become clinical useful.
With an increasingly extensive catalog of genetic lesions that MDS cells can harbor, a framework is emerging that helps explain how mutations cooperate to create the clonally expanded, dysplastic hematopoiesis characteristic of MDS. Genetic lesions contribute, to varying extents, to a survival advantage for abnormal hematopoietic stem cells over their normal counterparts or the acquisition of stem cell like self-renewal in a committed myeloid progenitor, dysplastic development of one or more cell lines, increased apoptosis of differentiating cells, or changes in genomic instability. Ultimately, mutations lead to the increase in proliferation and more complete block in differentiation associated with progression to acute myeloid leukemia.
This article focuses on our understanding of the genetic basis of MDS to date, with more attention given to recent discoveries in this field and the novel technologies that have made them possible.
Chromosomal amplifications and deletions
The large chromosomal deletions found in patients with MDS contain many protein coding genes and several less well-characterized noncoding genes. The large range of potential targets has made it difficult to identify the mechanisms by which gene loss leads to MDS. However, the search for MDS-associated genes has been helped by comparing chromosomal deletions across many patients. Mapping of the proximal and distal breakpoints from different patients has been used to create and narrow the boundaries of commonly deleted regions (CDRs). The long arm of chromosome 5 is the most common chromosomal deletion in MDS and has been found to contain two distinct CDRs. The more distal of these lies in 5q33.1 and was defined by analyzing deletions in patients with the clinical phenotype described by the 5q- syndrome – refractory anemia, thrombocythemia, and low risk of progression to AML. This CDR contains 40 protein coding genes, three known micro RNAs, and at least two lincRNA genes.
Because only one allele is deleted in 5q- MDS, initial efforts focused on the identification of genes that are inactivated on the intact allele, consistent with homozygous inactivation of a tumor suppressor. No mutations or microdeletions on the intact allele have been identified. An alternative hypothesis is that loss of a single allele, leading to a quantitative decline of its gene product, could contribute to the development of MDS.
By targeting each of the 40 known genes in the 5q- syndrome CDR with short-hairpin RNAs (shRNAs), we identified the ribosomal protein S14 ( RPS14 ) gene as a causal gene for the 5q- syndrome, capable of reproducing in vitro the profound block in erythroid differentiation seen in patients with this MDS variant. The introduction of an exogenous RPS14 gene rescued the block in erythroid differentiation in cultured CD34 + cells from 5q- syndrome MDS patients. Of note, congenital mutations in one allele of other ribosomal proteins have been identified in Diamond Blackfan Anemia, a rare disorder with a strikingly similar erythroid phenotype as the 5q- syndrome. The mechanism of erythroid failure appears to involve activation of the p53 pathway.
Most patients with 5q deletions do not have the clinical features of the 5q- syndrome and carry more proximal breakpoints that result in the loss of both 5q CDRs. The proximal common deleted region contains many genes, including CTNNA1, which is epigenetically silenced in some cases of 5q-, EGR1 , and HSPA9 . Heterozygous inactivation of EGR1 increases hematopoietic stem cell self-renewal, and may therefore contribute to clonal advantage of cells with deletion of this region. The full phenotype of 5q- MDS is likely caused by the integrated effects of haploinsufficiency for multiple genes as reviewed recently.
Several genes that lie outside of the 5q- CDRs have also been implicated in myeloid malignancies and are often lost in patients who have MDS with large 5q deletions. These genes include NPM1 and the tumor suppressor APC . NPM1 mutations are the most commonly identified mutations in patients with cytogenetically normal AML. Inactivation of one allele of NPM1 in a murine model results in an MDS-like phenotype.
Patients with low risk MDS and 5q- have a striking response to lenalidomide, a derivative of thalidomide, with 67% achieving transfusion independence and 45% achieving a cytogenetic response in a Phase II clinical trial. Two phosphatases, CDC25C and PP2A , are located on 5q and have been implicated in the response to lenalidomide. CDC25C is located within the proximal 5q CDR and is directly inhibited by lenalidomide in vitro. Knockdown of these phosphatases sensitizes cells to lenalidomide-mediated apoptosis. Similarly, the SPARC gene is located in the distal common deleted region, and treatment with lenalidomide increases SPARC gene expression.
Genes from other commonly deleted regions have not been as clearly implicated in the pathogenesis of MDS. Multiple CDRs on chromosome 7q have been described that include several candidate genes, but most patients with 7q deletions have lost two or more of these regions. Unlike the favorable prognosis associated with an isolated 5q deletion, the presence of any chromosome 7 abnormality, including uniparental disomy, has been shown to be a marker of poor outcomes in MDS. Which gene or genes are responsible for this negative prognosis have yet to be well characterized.
Isolated deletion of the long arm of chromosome 20 (20q-) is the third most common recurrent chromosomal loss in MDS after deletion of 5q and 7q. This abnormality is also found in AML and represents the most frequent chromosomal deletion in myeloproliferative neoplasms (MPN). The common deleted region of 20q has been mapped to a segment between 20q11 and 20q13. This smallest CDR contains 19 genes, none of which have been found to be mutated or otherwise directly implicated in myeloid diseases to date. Isolated loss of 20q is incorporated into the IPSS risk score and is considered a positive prognostic marker. However, there is evidence that late emergence of 20q- during disease progression is an indicator of genomic instability and may be associated with poor outcome.
Trisomy 8 (+8) is the only common chromosomal amplification recurrently associated with MDS. It can be found in other hematologic malignancies and in some solid tumors. Gene expression analyses of leukemic cells have shown increased transcription of genes from chromosome 8 from patients with trisomy 8 AML. Comparison of this expression pattern to that of normal CD34 + cells revealed upregulation of antiapoptotic genes. Similar studies with CD34 + cells from patients with MDS demonstrated increased expression of pro- and antiapoptotic genes. When tested in culture, trisomy 8 CD34 + cells were resistant to irradiation and were able to form hematopoietic colonies despite expression of apoptotic proteins normally associated with rapid senescence and cell death. The clinical correlate of this cellular phenotype is decreased survival seen in patients with +8 MDS (22 months mean survival compared with 53 months for normal karyotype MDS) and a lower response to chemotherapy in patients with +8 AML.
A subset of patients with MDS and trisomy 8 as their sole cytogenetic abnormality are notable for their clinical response to immune suppression. These patients tend to be younger, have refractory anemia of short duration, and are more likely to carry HLA DR15. Studies have shown expansion of Vβ-restricted CD8 + positive T-cells in patients with +8 MDS and a return to more normal polyclonal Vβ expression after successful treatment with immunosuppressants. Patients who respond to immunosuppression can achieve transfusion independence and even normalization of their red blood cell count. However, successful treatment does not abolish the +8 clone, which can actually expand in the bone marrow of responders after immunosuppression. These findings suggest that the +8 clone is resistant to killing by cytotoxic T-cells and likely has a growth advantage compared with normal hematopoietic precursors that are enhanced in the setting of autoimmunity. This immune mechanism may play a role in cases of MDS without +8, as the response to immunotherapy is not exclusive to this group.
Isolated loss of chromosome Y (-Y) in hematologic cells is an age-related phenomenon and most often is not associated with a myeloid disorder. In patients with MDS, loss of Y in the majority of cells can be a useful marker of disease burden, but likely has no bearing on the mechanism of disease. Like patients with a normal karyotype, those with -Y are considered to have prognostically favorable cytogenetics in the IPSS.
Balanced translocations
Analysis of gene partners in balanced translocations can be very informative, since it identifies genes likely to be involved in the pathogenesis of the underlying disease. This has been a particularly fruitful approach in chronic and acute myeloid leukemias. However, unlike AML, recurrent chromosomal translocations in MDS are rare. Recurrent translocations do occur involving the MDS1-EVI1 locus on chromosome 3q and between partners on chromosomes 6p and 9q. Other genes that are involved in rarer translocations include RUNX1 , TEL , MEL1 , NUP98 , and IER3 . Many translocations in MDS occur in the setting of a more widely disturbed cytogenetic profile, making it unclear if these represent disease modifying changes or incidental rearrangements in a highly unstable genome.
The MDS1-EVI1 gene locus is unusual in that it encodes two related proteins with partially opposing functions. Alternative splicing of transcripts results in either expression of the full length MDS1-EVI1 gene product or just the Evi1 protein alone. Evi1 can repress the function of Gata1 and thus alter normal hematopoietic differentiation. It can also down-regulate activity of the Mds1-Evi1 fusion protein. The MDS1-EVI1 locus is one of the most common translocation partners in myeloid malignancies and has been identified as a common site for retroviral insertional oncogenesis. Presence of a 3q translocation is typically associated with a poor prognosis. In patients with MDS or AML without a 3q translocation, overexpression of EVI1 often occurs and also confers a poor prognosis. Finally, EVI1 overexpression in a mouse bone marrow transplant model generates an MDS like phenotype. Although translocations involving this locus on 3q are typically associated with AML, they are frequent enough in MDS that they can be used as evidence to support a diagnosis of MDS in patients with equivocal evidence of dysplasia.
Translocations between chromosomes 6p and 9q are also considered presumptive evidence of MDS in patients with unexplained anemia and little evidence of marrow dysplasia. In AML, where this translocation is more common, it is a marker of poor prognosis. The genes involved in this translocation are DEK (on 6p21) and NUP214 on 9q34. DEK encodes a DNA binding protein and can inhibit histone acyltransferase activity and modulate transcript splicing. NUP214 encodes a member of the nuclear pore complex. The DEK/NUP214 fusion protein up-regulates protein synthesis in myeloid cells, but how it might contribute to leukemogenesis or dysplasia is not yet clear.
A novel MDS-related gene was recently discovered because of its involvement in a slightly different 6p to 9q translocation. In this case, the IER3 gene from 6p21 was fused to a transcript-poor region of 9q34, resulting is loss of expression of the IER3 gene product. IER3 is an immediate early response gene involved in proapoptotic signaling. Evidence for IER3 rearrangements was found in several other patients and expression of IER3 was dysregulated in more than half of patients who have MDS without 6p abnormalities.
Balanced translocations
Analysis of gene partners in balanced translocations can be very informative, since it identifies genes likely to be involved in the pathogenesis of the underlying disease. This has been a particularly fruitful approach in chronic and acute myeloid leukemias. However, unlike AML, recurrent chromosomal translocations in MDS are rare. Recurrent translocations do occur involving the MDS1-EVI1 locus on chromosome 3q and between partners on chromosomes 6p and 9q. Other genes that are involved in rarer translocations include RUNX1 , TEL , MEL1 , NUP98 , and IER3 . Many translocations in MDS occur in the setting of a more widely disturbed cytogenetic profile, making it unclear if these represent disease modifying changes or incidental rearrangements in a highly unstable genome.
The MDS1-EVI1 gene locus is unusual in that it encodes two related proteins with partially opposing functions. Alternative splicing of transcripts results in either expression of the full length MDS1-EVI1 gene product or just the Evi1 protein alone. Evi1 can repress the function of Gata1 and thus alter normal hematopoietic differentiation. It can also down-regulate activity of the Mds1-Evi1 fusion protein. The MDS1-EVI1 locus is one of the most common translocation partners in myeloid malignancies and has been identified as a common site for retroviral insertional oncogenesis. Presence of a 3q translocation is typically associated with a poor prognosis. In patients with MDS or AML without a 3q translocation, overexpression of EVI1 often occurs and also confers a poor prognosis. Finally, EVI1 overexpression in a mouse bone marrow transplant model generates an MDS like phenotype. Although translocations involving this locus on 3q are typically associated with AML, they are frequent enough in MDS that they can be used as evidence to support a diagnosis of MDS in patients with equivocal evidence of dysplasia.
Translocations between chromosomes 6p and 9q are also considered presumptive evidence of MDS in patients with unexplained anemia and little evidence of marrow dysplasia. In AML, where this translocation is more common, it is a marker of poor prognosis. The genes involved in this translocation are DEK (on 6p21) and NUP214 on 9q34. DEK encodes a DNA binding protein and can inhibit histone acyltransferase activity and modulate transcript splicing. NUP214 encodes a member of the nuclear pore complex. The DEK/NUP214 fusion protein up-regulates protein synthesis in myeloid cells, but how it might contribute to leukemogenesis or dysplasia is not yet clear.
A novel MDS-related gene was recently discovered because of its involvement in a slightly different 6p to 9q translocation. In this case, the IER3 gene from 6p21 was fused to a transcript-poor region of 9q34, resulting is loss of expression of the IER3 gene product. IER3 is an immediate early response gene involved in proapoptotic signaling. Evidence for IER3 rearrangements was found in several other patients and expression of IER3 was dysregulated in more than half of patients who have MDS without 6p abnormalities.
Uniparental disomy and copy number variation
Several recently developed technologies are capable of genome-wide assessment of DNA copy number, identifying amplifications and deletions in an unbiased manner that are not apparent by standard cytogenetic analysis. These techniques include high-density comparative genome hybridization (aCGH), single nucleotide polymorphism (SNP) microarrays and copy number variation (CNV) genotyping, and next-generation DNA resequencing technologies. Genome-wide sequencing using next-generation technologies has not yet been applied to MDS, although aCGH and SNP array studies have been performed extensively. These studies have already highlighted novel MDS disease alleles and loci.
SNP microarrays were initially designed to genotype single nucleotide polymorphisms, but they can also detect changes in DNA copy number, including small chromosomal deletions or amplifications, that are unrecognized by traditional karyotyping. A commonly used version of these chips, the Affymetrix 6.0 array, has probes dedicated solely to the determination of copy number at nearly one million loci on the human genome. Areas of acquired copy number neutral, uniparental disomy (aUPD) are also detectable with these instruments and can point to disease-related abnormalities that would otherwise go unnoticed. Several groups have used SNP-arrays to examine DNA from MDS bone marrow samples. One of their common findings is that more than half of the patients who have MDS with normal cytogenetics by metaphase karyotyping harbor cryptic copy number changes or aUPD. Some of these changes are microdeletions or aUPD at the loci of known MDS karyotypic abnormalities, such as on the long arm of chromosome 7. More importantly, these findings appear to have prognostic importance in MDS and AML. In addition, SNP array studies have highlighted genes that were subsequently found to be mutated in a substantial portion of cases, including the TET2 gene and CBL described below.
Point mutations in myelodysplastic syndrome genes
TET2 Mutations
The TET1 gene was identified as a rare translocation partner with the mixed-lineage leukemia gene (MLL) in a fraction of patients with AML. Neither this gene, nor the other two genes it shares homology with, TET2 and TET3 , had previously been associated with malignancy. In the last year, TET2 was reported to be mutated at high frequency in several myeloid malignancies, including MDS. Using SNP-arrays, analysis of DNA samples from patients with MDS, MDS/MPN, and AML revealed recurrent acquired uniparental disomy and small deletions in chromosomal segment 4q24. The only open reading frame in the smallest commonly altered region was the TET2 gene and mutations of highly conserved codons in TET2 were identified in these patients. TET2 mutations were not limited to a single class of myeloid disorders. Many patients with MPN (∼10%) had TET2 mutations (often associated with loss of heterozygosity of 4q24) as did a large fraction of unselected patients with MDS (∼20%), chronic myelomonocytic leukemia (CMML) (∼30%), and secondary AML (∼25%), including those without obvious alterations of the 4q24 locus. Subsequent studies have confirmed that TET2 mutations are not exclusive of several genetic abnormalities and are found alongside PML-RAR , FLT3-ITD , JAK2 V617F , MPL W515L/K , KIT D816V , NRAS , and NPM1 mutations and in myeloid malignancies with no known mutations. Point mutations of TET1 or TET3 were not found in a large set of MPN samples and have not been reported in other myeloid malignancies to date.
No other recurring gene mutation has been identified in such a broad spectrum of myeloid neoplasms at such high frequency, which prompts the speculation that impairment of TET2 function promotes a common element in the pathogenesis of all of these diseases, namely the establishment or maintenance of clonal hematopoiesis. Mutations of this gene could not be the sole cause of the dysplasia seen in patients with MDS because patients with polycythemia vera (PV) carrying TET2 mutations have normal (albeit amplified) erythroid differentiation. Nor does TET2 seem to promote transformation to acute leukemia in isolation. In a study of 96 subjects with MDS, TET2 mutations were no more likely to be found in those with high risk disease nor were any new mutations seen during disease progression. In another study, TET2 mutations were found more frequently in low and intermediate-1 risk groups.
Instead, it appears that impairment of TET2 function may provide a survival advantage to early hematopoietic progenitors without profound effects on differentiation or proliferation. Single colony assays of CD34 + cells from the bone marrow of patients with JAK2 V617F and TET2 mutations support this view. A subset of more primitive CD34 + CD38 − cells from these patients often carried TET2 mutations without associated JAK2 mutations. The proportion of TET2 -only mutant cells was greater in this CD34 + CD38 − group than in more mature CD34 + CD38 + cells. In the five subjects studied, no cells carrying the JAK2 V617F mutation were found to have wild-type TET2 . CD34+ stem cells from two subjects with TET2 mutated/ JAK2 V617F positive neoplasms could repopulate the bone marrow of sub-lethally irradiated NOD-SCID mice, whereas cells from three subjects with the JAK2 mutation and wild-type TET2 could not. Fifteen weeks after engraftment, single colony assays of surviving cells revealed that the population of TET2 mutated, JAK2 wild-type cells had grown at the expense of those cells carrying mutations in both genes. This finding suggests that the TET2 mutation might have preceded the JAK2 V617F mutation in these subjects and that it resides in a more primitive hematopoietic stem cell.
Evidence for mutations in TET2 as an early event in myeloid disorders is not universal, however. Saint-Martin and colleagues studied four family cohorts with a strong inherited predisposition for MPNs and AML. These subjects did not carry germline JAK2 mutations, but often acquired them when they presented with myeloid malignancies. Many of the affected members of these cohorts developed TET2 mutations, but several others did not, and no two related subjects acquired the same TET2 mutation. In one subject with JAK2 V617F / TET2 -mutant PV for which several bone marrow samples were available, it was noted that the subject had mostly JAK2 V617F -positive clones that were TET2 negative at an early stage of the disease. The remaining clones were either completely wild-type or contained the JAK2 V617F mutation and a single TET2 mutation. When the subject progressed to acute leukemia, almost all bone marrow mononuclear cells contained biallelic TET2 mutations in addition to JAK2 V617F . These findings demonstrate that TET2 mutations can be either primary or secondary events and are consistent with impaired TET2 function creating a selective growth advantage for disease stem cells.
Less is known about the role of TET2 mutations in MDS. In a study of 96 subjects with MDS, Kosmider and colleagues found mutation of TET2 to be a positive prognostic factor independent of IPSS risk group. There was no difference in clinical markers at the time of presentation in the mutated compared with the non-mutated group. In contrast, other groups have found that patients with AML or CMML that carry TET2 mutations have decreased overall survival. It is not clear why TET2 mutations appear to be protective in MDS, but are unfavorable in more proliferative myeloid diseases. It is possible that patients who have MDS with TET2 mutations as the basis of their disease lack other, less favorable genetic lesions, and hence have better outcomes. But for the subset of patients with more aggressive diseases, the lack of TET2 function could enhance the effect of the additional mutations they have acquired or impart a relative resistance to treatment. Studies with a much larger number of subjects are needed to better understand the prognostic implications of TET2 mutations.
The mechanism through which TET2 mutations promote diverse myeloid malignancies is not well understood. However, a recent study on the function of its homolog TET1 suggests that TET2 may affect epigenetic modulation of gene expression. TET1 encodes a 2-oxoglutarate and Fe(II)-dependent enzyme that catalyzes the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC). This modified base was found to comprise roughly 5% of all CpG islands in mouse embryonic stem cells. The authors speculate that 5-hmC could be a chemical intermediate in the demethylation of cytosine residues, either by spontaneous or enzyme-mediated conversion. Alternatively, DNA methyltransferases may not recognize 5-hmC, and therefore, may not methylate cytosines on newly synthesized DNA strands complementary to regions with these epigenetic marks. The result would be a net loss of cytosine methylation mediated by TET proteins. MDS and AML are disorders whose pathogenesis involves hypermethylation of several tumor suppressor genes, and are diseases for which inhibitors of DNA methyltransferases have shown efficacy. It is intriguing to have identified a frequently mutated gene whose likely mechanism of action could explain the basis for these observations. Of particular interest will be the analysis of whether TET2 -mutation status predicts response to therapy with the hypomethylating agents 5-azacytidine and decitabine.
ASXL1 Mutations
The ASXL1 gene is another example of a gene that has recently been found to carry mutations in a variety of myeloid disorders including MDS. This gene encodes a chromatin-binding protein that functions as a ligand-dependent coactivator of the retinoic acid receptor. It regulates the response to retinoic acid in a cell-type specific manner and is thought to mediate its effects through a direct interaction with the histone acyltransferase NCOA1 (also known as SRC-1 ) or the histone demethylase LSD1 . ASXL1 is located in 20q11, but is proximal to the 20q common deleted region in patients with MDS.
ASXL1 was identified as a myeloid disease gene from aCGH experiments analyzing germline and CD34 + -cell DNA from a small set of patients with MDS/AML. One individual had a small, atypical deletion on chromosome 20 that encompassed the ASXL1 locus. Subsequent sequencing identified mutations of this gene in 16% of subjects with MDS/AML and in 43% with CMML. Most of these samples were studied to see if they carried mutations in several other MDS associated genes, including HRAS , KRAS , NRAS , RUNX1 , NFIA , CTNNB1 , and TET2 . In this small sample set, mutations in RUNX1 and TET2 were identified in subjects with MDS and these did not coexist with ASXL1 mutations. In CMML cases, RUNX1 and TET2 mutations were not exclusive of ASXL1 mutations. In contrast, ASXL1 mutations were found in several MPN samples, but none of these were from subjects with PV and none carried the JAK2 V617F mutation. This finding suggests that loss of ASXL1 activity might replace the pathogenicity of the constitutively activated JAK2 oncogene in a subset of JAK2 wild-type myeloproliferative diseases. As was the case with CMML samples, TET2 mutations were not exclusive of ASXL1 mutations in these MPNs. Mutations in ASXL1 were also identified in 20% of a group of subjects with AML, most of which had normal cytogenetics. Although nearly half of these subjects had NPM1 mutations, ASXL1 and NPM1 mutations were epistatic. In this study, the investigators suggest that this mutual exclusivity could mean that ASXL1 and NPM1 contribute to the same pathogenic pathway. The alternative interpretation is that these genes represent completely independent mechanisms of transformation because many of the ASXL1 mutated AML cases arose from chronic myeloid diseases, whereas most NPM1 -mutated cases did not.
Exactly how ASXL1 contributes to the development of MDS and other myeloid disorders is not clear. ASXL1 deficient mice do not have a dramatic myeloerythroid phenotype or gross changes in their stem cell compartment. The higher incidence of ASXL1 mutations in CMML and AML compared with MDS and its mutual exclusivity with JAK2 in MPNs prompts the speculation that ASXL1 mutations may drive cell proliferation with few effects on morphology or differentiation. Finally, the clinical implications of ASXL1 mutations in patients with MDS are not yet known.
CBL Mutations
The CBL gene encodes an E3 ubiquitin ligase and contains a tyrosine kinase binding domain. It negatively regulates tyrosine kinase activity by promoting ubiquitination of these proteins, leading to their degradation. Using the same approach that led to the discovery of TET2 mutations, mutations of the CBL gene were found in a large fraction of patients with myeloid disorders who were noted to have acquired uniparental disomy of 11q. Most of these patients had CMML or atypical chronic myelocytic leukemia (aCML), but CBL mutations were found in a few patients with refractory anemia with excess blasts. In several CBL mutants without 11q UPD, a second inactivating mutation in CBL was identified. Although some mutations appear to cause loss of function, other evidence indicates that CBL may function as an oncogene. Mutations in CBL occur almost exclusively in a short segment of the protein coding region and maintain the reading frame of the gene. The mutated protein product has impaired ubiquitin ligase activity and can impair the function of wild-type CBL in a dominant negative fashion. Expression of mutant CBL in 3T3 cells enhances their ability to form colonies in agar and tumors in nude mice. In this sense, CBL appears to be an oncogene and not a tumor suppressor. The answer is that it may be a little of each. Mice deficient for cbl have an expanded hematopoietic progenitor pool and some degree of hypersensitivity to cytokine signaling, but do not develop MDS or MPN. When a mutant CBL gene is inserted into hematopoietic cells from these mice, they show dramatic hypersensitivity to cytokine stimulation and proliferate much more. It appears that CBL mutants gain the ability to promote tyrosine kinase signaling that is separate from their loss of ubiquitin ligase activity, possibly through dominant negative inhibition of its homolog CBLB .
CBL mutations have previously been identified in rare cases of de novo AML, and are most frequently associated with core-binding factor leukemias. Most CBL mutant samples from patients with AML are negative for mutations in FLT3 , KIT , NPM1 , CEBPA , and RAS genes, implying that loss of CBL function can substitute for the growth advantage conveyed by these oncogenic mutations more commonly associated with AML. CBL mutations are also found at high frequency (17%) in cases of juvenile myelomonocytic leukemia (JMML). These mutations are exclusive of RAS and PTPN11 mutations, which are associated with 25% to 35% of JMML cases.
Although CBL mutations have been found in patients with MDS, it is primarily associated with the monocytic myeloproliferative disorders JMML, CMML, and aCML. Its mechanism of action and lack of association with other pro-proliferative oncogene mutations suggest that mutation of CBL has an effect comparable to that caused by constitutive activation of tyrosine kinases. It remains to be seen whether CBL mutations play a role in lower risk MDS or whether loss of this protein function confers a worse prognosis in this disease.
Receptor Tyrosine Kinase and RAS Pathway Mutations
Activating mutations in the RAS family of oncogenes were among the first frequently recurring point mutations identified in patients with MDS. They are present in roughly 15% of MDS cases and are associated with an increased risk for progression to AML. Various missense mutations in codon 12 of NRAS are the most common RAS mutations in MDS, with mutations at other sites or in KRAS occurring at lower frequency. Like CBL mutations, oncogenic RAS mutations are more common in patients with CMML (∼50%) and JMML (∼30%).
RAS proteins are mediators of signal pathways downstream of receptor tyrosine kinases, such as those that mediate cytokine sensitivity in hematopoietic cells. These include receptors for thrombopoietin, erythropoietin, stem cell factor, Flt-3 ligand, platelet-derived growth factors, several interleukins, insulin and insulin-like growth factors, and the colony stimulating factors. Some of these receptors have been found to have activating mutations in advanced cases of MDS or CMML, including FLT3 , CSF1R (aka c-FMS ), and KIT , although these are more commonly associated with AML. Other RTK-RAS pathway genes mutated in myeloid disorders include BRAF and PTPN11 , both of which are rare in MDS.
TP53 Mutations
Mutations of the critical cell-cycle checkpoint gene TP53 are among the most common genetic abnormalities in malignancies. In MDS, TP53 mutations occur in 10% to 15% of patients, but this frequency can be higher in those with prior exposure to alkylating agents or radiation. Inactivating mutations of TP53 are associated with advanced disease, complex karyotype, and resistance to treatment; all of which portend a poor prognosis. The presence of TP53 mutations may even be an independent risk factor after accounting for IPSS risk score. As the sensitivity of molecular diagnostics improve, it will be feasible to determine if patients carry TP53 mutations in diseased subclones at low frequency. This treatment resistant TP53 -mutant population may drive progression of disease and expand after therapy even in patients who otherwise appear to have a good prognostic profile.
RUNX1 Mutations
RUNX1 is a member of the core-binding factor family of proteins and an important regulator of normal hematopoiesis. Like TET2 , RUNX1 abnormalities are found in many myeloid disorders. In MDS, mutations are present in 7% to 15% of cases of de novo disease, but appear to be more frequent in therapy-related MDS. Patients who have MDS with RUNX1 mutations are more likely to have advanced disease and carry a poor prognosis.
The RUNX1 protein contains two primary domains important for its function: a proximal Runt DNA binding domain and a distal activation domain. Mutations are found throughout the length of the gene in patients with de novo MDS, but tend to cluster in the DNA binding domain in patients with AML and therapy-related MDS. Heterozygous Runt domain mutations can act as dominant negatives, severely limiting RUNX1 activity, and may be partially responsible for the poorer outcome of patients with therapy-related disease.
Manipulation of RUNX1 in mice has helped explain how acquired RUNX1 mutations could lead to MDS and AML. Complete RUNX1 knockout results in embryonic lethality without evidence of definitive hematopoiesis. However, selective RUNX1 excision in adult animals does not block hematopoiesis. Instead, these mice show lymphoid defects, expansion of the myeloid progenitor pool, inefficient platelet production, and extramedullary hematopoiesis, but not progression to AML. In contrast, mice transplanted with bone marrow cells overexpressing RUNX1 with a Runt domain mutation developed increasing bone marrow blast counts, splenomegaly, and often died of an AML-like disease. Mice transplanted with cells expressing RUNX1 with a more distal frameshift mutation instead developed a more MDS-like phenotype with marked erythroid dysplasia and pancytopenia with a lower rate of progression to AML.
Further evidence of the role of RUNX1 in myelodysplasia and progression to AML comes from the rare autosomal dominant disease known as the familial platelet disorder with propensity to acute leukemia (FPD). Affected persons with FPD often have an MDS-like disease characterized by low number of platelets that may function abnormally. About 35% of patients with FPD will develop AML, often by acquiring a second RUNX1 mutation in their remaining wild-type allele.
In the mouse models and human cases of congenital RUNX1 mutations, progression of disease occurs only after a long latency and likely requires cooperating mutations. In patients with MDS and patients with AML arising from MDS, RUNX1 mutations have been associated with activating mutations in the receptor tyrosine kinase-RAS pathway and the presence of chromosome 7 deletions, which suggests that these abnormalities are complementary, each driving a distinct step in the pathogenesis of more advanced disease.
JAK2 Mutations
Activating mutations of JAK2 are the defining genetic lesion of PV and are found in roughly half of patients with essential thrombocythemia (ET) or primary myelofibrosis. In MDS, the mutation rate is much lower with the JAK2 V617F mutation reported in approximately 5% of unselected patients with MDS. This frequency is higher in a subset of patients. More than half of patients with refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T) carry the JAK2 V617F mutation and a smaller number contain activating mutations of MPL . These patients typically have low risk disease and rarely progress to acute leukemia. This mutation frequency is comparable to that seen in essential thrombocytosis, prompting the speculation that some cases of RARS-T could be considered to be the frustrated expression of a myeloproliferative disorder and not always a subcategory of MDS. The alternative view is that the acquisition of a JAK2 V617F mutation in the setting of RARS leads to thrombocytosis, but patients retain the risk of progression associated with RARS.
There remains a population of JAK2 V617F positive patients who have MDS without ringed sideroblasts. Whether these patients have an MDS/MPN overlap syndrome or simply MDS is less clear. Analysis of a 5q- MDS cohort demonstrated the JAK2 mutation in 6% of these patients. Those positive for JAK2 V617F had higher peripheral white blood cell counts, a trend toward a higher platelet count, but no excess blasts in their bone marrow. One patient sample was subject to clonal analysis, identifying the 5q- abnormality in 91% of metaphases. However, single colony assays revealed the JAK2 V617F mutation in closer to 50% of cells, which suggests that the JAK2 mutation developed in a subclone that had already acquired the 5q- deletion. In this genetic background, constitutive JAK2 activity may provide a clonal advantage without being able to drive the proliferation of terminally differentiated cells seen in PV and ET. It also suggests that targeting JAK2 activity in patients who have MDS with JAK2 V617F mutations might do little to ameliorate the risk of subsequent disease progression.
Related posts:
 The Epidemiology of Myelodysplastic Syndromes
Mouse Models of Myelodysplastic Syndromes
Hematopoietic Stem Cell Transplantation for MDS
The Epidemiology of Myelodysplastic Syndromes
Mouse Models of Myelodysplastic Syndromes
Hematopoietic Stem Cell Transplantation for MDS
 Lenalidomide for Treatment of Myelodysplastic Syndromes: Current Status and Future Directions
Novel Therapies for Myelodysplastic Syndromes
Lenalidomide for Treatment of Myelodysplastic Syndromes: Current Status and Future Directions
Novel Therapies for Myelodysplastic Syndromes
 Patient Selection for Transplantation in the Myelodysplastic Syndromes
Patient Selection for Transplantation in the Myelodysplastic Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


