Keywords
FDA, CBER, cancer gene therapy, regulatory review, CMC, pharmacology/toxicology, clinical, drug development
Introduction
FDA’s Regulatory Responsibility and Gene Therapy Submissions
In the United States, the U.S. Food and Drug Administration (FDA) is responsible for the regulation of clinical trial research using investigational products, including gene therapies for cancer indications. FDA’s authority and regulatory processes relating to drugs, biologics, and medical devices are covered in other current reviews . Human gene therapies, along with other biologic therapeutics, are regulated within FDA by the Center for Biologics Evaluation and Research (CBER).
The first human gene therapy trial was conducted in 1990 at the National Institutes of Health Clinical Center in a patient with adenosine deaminase deficiency . The first clinical trial to use gene therapy to treat cancer followed soon thereafter . Since submission of the first gene therapy Investigational New Drug (IND) application in 1989, 754 human gene therapy INDs have been submitted to CBER for review (through 2011), of which 563 have been for cancer indications ( Figure 35.1 ). At the end of 2011, 333 gene therapy INDs remained active, 256 of which were for cancer indications.
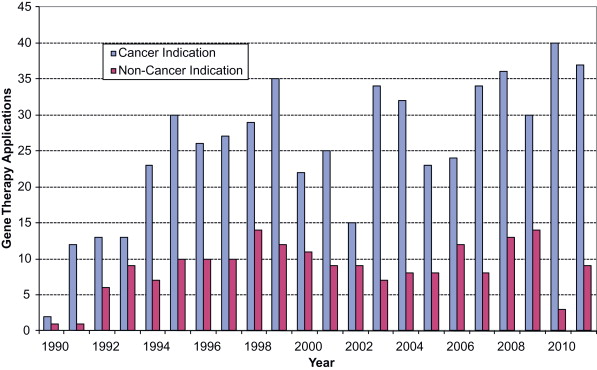
After the initial gene therapy investigations in 1990, there was a rapid increase in gene therapy INDs submitted to FDA, mostly for cancer indications.
Overview of CBER’s IND Review Process
The review of gene therapy INDs is coordinated through the Office of Cellular, Tissue, and Gene Therapies (OCTGT) within CBER. No human gene therapy has yet been approved for sale in the United States. In contrast to cytotoxic drugs and monoclonal antibody products that are typically developed by large pharmaceutical companies, gene therapies are often developed in academic centers and smaller biotechnology companies. Individuals, academic institutions, companies, or other organizations that take responsibility to initiate a clinical investigation of a new product are referred to as sponsors. An active IND application allows a sponsor to initiate clinical research.
Interactions between sponsors and CBER can be highly collaborative. CBER regulatory and scientific staff work closely with gene therapy sponsors, clinicians, and scientists at all stages of product development. Although interactions between CBER and gene therapy sponsors may commence at the time of submission of an IND, CBER offers a pre-investigational new drug application (pre-IND) consultation program to foster early communications between sponsors and CBER . During these pre-IND meetings, the sponsor is able to present relevant preliminary preclinical data, manufacturing data and plans, and a proposed clinical development scheme. During this interaction, specific questions may be asked of CBER experts, who can provide sponsors with regulatory, scientific, and clinical responses to permit a more efficient development program for their product.
The types of information that an IND submission must contain are detailed in the Code of Federal Regulations , Section 312.23 . IND sections that require detailed CBER review include the investigational product’s chemistry, manufacturing, and control (CMC) data; preclinical information on the pharmacology and toxicology assessment or pertinent scientific references; and the clinical protocol. FDA’s mandated role is to determine if the clinical trial design is acceptable to ensure the safety of subjects and has the potential to attain the scientific objectives of the investigation.
The initial FDA review of a new IND will occur within 30 days . At the conclusion of the 30-day review clock, there are two possible outcomes:
- 1.
The IND becomes active, and the protocol may proceed.
- 2.
The IND will be placed on hold for deficiencies that the Agency must clearly specify.
It is important to note that FDA neither formally approves nor endorses an IND study. Rather, if CBER’s review team determines that the IND submission is satisfactory and the clinical study is acceptable in terms of patient safety and scientific merit, then the IND becomes active at the end of the 30-day review, and the investigator(s) may commence the trial. However, if a clinical hold is imposed, FDA will provide the sponsor with a clinical hold letter that lists specific deficiencies to be addressed prior to initiation of the clinical investigation. The sponsor must address all hold issues and submit revisions as an amendment to the IND, which FDA will review within 30 days. The IND submission and review process is depicted in Figure 35.2 .
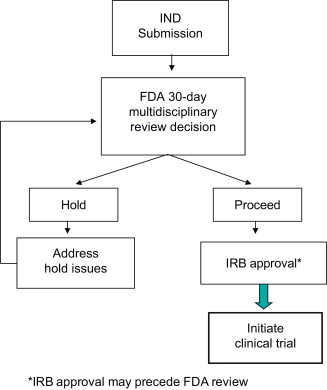
The remainder of this chapter focuses on CBER’s multidisciplinary review process for cancer gene therapy INDs and products.
CMC Review
Classes of Cancer Gene Therapy Products
Cancer gene therapies encompass a diverse array of products that may include plasmids, viral vectors, bacterial vectors, and genetically modified human cells. Because each class of cancer gene therapy product is confronted by unique issues during manufacturing and testing, it is useful to outline the major categories of these products before proceeding further:
- 1.
Nonreplicating gene therapy vectors for direct in vivo use : Nonreplicating gene therapy products express one or more foreign transgenes or RNA transcripts from a vector. The vector may be derived from a plasmid, virus, or microbial cell. In some cancer gene therapies, the foreign transgene may have direct or indirect antitumor activity (prodrug-converting, proinflammatory, anti-angiogenic, cytocidal, etc.). In other cases, the transgene may be a tumor antigen that is intended to induce a specific immune response (tumor vaccine).
- 2.
Replication-competent gene therapy vectors: A number of viruses are able to replicate particularly well in transformed cells. Such viruses may be further engineered to enhance their ability to replicate in tumor cells or to disable replication in nontumor cells. These products are often referred to as oncolytic viruses. Such viruses are considered gene therapies if they also express a foreign transgene. Many of the manufacturing and testing considerations are similar regardless of whether or not the oncolytic virus includes a foreign transgene.
- 3.
Cell therapies that are genetically modified ex vivo : Gene therapy vectors may be used to temporarily or permanently modify the genome of cultured human cells, and these cells can then be administered to the subject. For example, T cells may be transduced with a tumor-specific T cell receptor, or antigen-presenting cells may be engineered to express a tumor antigen as a vaccine. In some cases, gene therapy vectors may be used solely during the manufacturing of cell therapies, with the gene therapy vector being absent from the final product.
Combination Products
Sometimes a cancer gene therapy product is administered in conjunction with a device or drug: FDA classifies these as combination products . IND sponsors need to carefully consider how the various components of a combination product interact with each other and how best to coordinate the development of each component during clinical trials .
As one example of a combination product, some biologics must be administered using specialized delivery devices such as a catheter or infusion pump. In this situation, the sponsor of the gene therapy product is usually not the manufacturer of the device, and therefore the sponsor of the gene therapy product may need to coordinate with the device manufacturer to obtain sufficient information to allow FDA review. Because some biologics may be adsorbed or inactivated by certain types of materials, it is important to evaluate the stability of the biologic in the device. A pre-IND meeting is an excellent opportunity for a sponsor to ask FDA what kind of information will be needed about the device.
Another example of a combination product can occur when a biologic is narrowly tailored to target a specific tumor pathway (an example of “personalized medicine”). A diagnostic assay will typically be needed to determine tumor status. Such an assay may involve genotyping the tumor or measuring the level of expression of specific biomarkers in the tumor or blood. This strategy requires having an accurate assay, possibly even during the very earliest stages of a phase I clinical trial. FDA’s Center for Devices and Radiological Health (CDRH) regulates these assays as in vitro diagnostic devices. Before initiating phase III trials, IND sponsors will need to discuss the adequacy of their diagnostic assay with CDRH.
If a device is essential for a cancer gene therapy, it is advantageous to coordinate product development with the device manufacturer and to discuss development of the device with FDA. In addition, the sponsor of the gene therapy has an interest in ensuring that the device is properly developed along with the biologic during clinical trials, and the sponsor needs to be aware of any plans by the manufacturer to change or discontinue the device.
Manufacturing and Testing
FDA has published numerous guidances and reviews on manufacturing and testing of biologics, including gene therapies, and the reader is referred to these documents for comprehensive information on these topics . Because of space limitations, we focus on those manufacturing and testing issues that may be of particular interest to sponsors of early stage cancer gene therapies or issues that may require special attention with certain cancer gene therapy products.
Good Manufacturing Practices
Adhering to current good manufacturing practices (cGMPs) is important at all stages of clinical development because it improves manufacturing reliability and product consistency and ultimately facilitates development of a safe and effective licensed product. However, FDA recognizes that full compliance with cGMPs is sometimes impractical or even impossible during the early stages of clinical development. This is especially true when the product in question is being manufactured at small scale in a single batch or when the product is a novel biologic that presents unique difficulties for manufacturing or testing. FDA therefore takes a flexible approach to cGMPs for products used in phase I trials .
This flexibility does not mean that cGMPs can be ignored for early phase products; rather, FDA asks sponsors to focus on those aspects of the cGMPs that are most important for product safety. Compliance with cGMPs during phase I investigations should always include written documentation of manufacturing and quality control procedures, as well as documentation of manufacturing and testing data. The manufacturer should also have adequate control over the manufacturing environment, including equipment, personnel, raw materials, and cleaning and changeover procedures. All of these areas require attention, even when manufacturing products for phase I trials, because they help to ensure the safety of the product.
Other aspects of cGMP, most notably validation of manufacturing processes and testing procedures, may be more appropriate when manufacturing products for later-phase trials. IND sponsors typically refine test specifications and validate assays during the course of clinical trials, and they may even develop entirely new assays. For example, licensed biologics require a potency assay that measures the biological function of the product. However, potency assays are often unique for each product, and designing an effective assay may require a considerable amount of research and development. Rather than requiring that a potency assay be established at the very beginning of phase I trials, FDA expects that a potency assay will be established and have appropriate release specifications before phase III trials begin. FDA has published a detailed guidance on potency assays for cellular and gene therapy products .
Many sponsors outsource production and/or testing of their cancer gene therapy product to contract manufacturing and testing companies, and this can be a cost-effective way to generate a product for clinical trials. The sponsor should keep in mind, however, that it is the sponsor, not the contractor, who bears ultimate responsibility for the quality of the product. Sponsors should always have a formal written quality assurance program in place, even at the earliest stages of clinical development, and even if the sponsor does not directly perform any in-house manufacturing or testing. This quality assurance program should be documented in the IND submission and should describe the responsibilities and qualifications of the sponsor’s quality control personnel, including responsibility for releasing product lots, approving manufacturing and testing procedures, and investigating and correcting deficiencies.
Product Changes and Comparability
Some degree of change in manufacturing processes is inevitable during the course of clinical development, including common changes such as scale-up and moves to new manufacturing sites. Other types of changes may be more avoidable, however, including major changes to the production cells, vector design, or purification processes. After a manufacturing change, sponsors need to demonstrate that a product remains functionally equivalent (comparable) and that the manufacturing change has not fundamentally altered the product . Comparability is typically demonstrated using analytical assays. However, complex products such as cell and gene therapies can be difficult to fully characterize with analytical assays, especially when the product’s mechanism of action may not be fully understood. Demonstrating product comparability can be an expensive and time-consuming exercise, and in extreme cases, it may require additional preclinical or clinical studies.
For these reasons, experienced manufacturers avoid major changes in product manufacturing once late-phase trials have begun. However, even for a drug developer who is at the very start of a product development program, it is worthwhile to think about ways to minimize the need for future changes in manufacturing. With biologics, one example of a common manufacturing change is a decision to avoid the use of animal-derived reagents (e.g., serum and trypsin) because of concerns about adventitious agents, most notably viruses and transmissible spongiform encephalopathy. Avoiding potential sources of contamination can be especially beneficial when the product is a replication-competent virus because such products can sometimes be very difficult to test for adventitious agents (discussed later). Although the move to animal-free reagents can be made at any stage during product development, it is often less complicated to design the initial manufacturing process to achieve this goal rather than waiting until later in the development process when such changes may trigger a need for comparability studies.
In general, changes in downstream processing (e.g., product purification) and changes in formulation tend to have more impact on the product than changes in upstream processing (e.g., culture conditions). For example, some small-scale purification processes for gene therapy vectors rely on ultracentrifugation, but larger-scale commercial processes may use chromatography. Such a major change in the manufacturing process may lead to substantial differences in product impurity profiles that require comparability studies and extensive investigation.
Sponsors are also well advised to design their manufacturing process with scalability in mind. Certain processes, such as ultracentrifugation, may be inherently difficult to scale up. As another example, transient transfection methods may be suitable for producing small lots of product for initial clinical studies but may become too unwieldy, labor-intensive, or variable at larger scales. If it is possible to manufacture a product using stably transfected cell lines, a decision to do this even at the earliest phases of clinical development will smooth the way to phase III trials. In summary, if one is intending to develop a product that could eventually be evaluated in late-phase trials, it will be advantageous to start with manufacturing and testing processes that are designed with this goal in mind.
Testing Vectors for Replication-Competent Contaminants
Contamination of a nonreplicating viral vector product with a wild-type revertant virus may present a serious safety concern. These replication-competent contaminants may arise due to homologous or nonhomologous recombination during manufacturing. Because even low-level contamination may pose a risk to subjects, it is important that assays for such contaminants be as sensitive as possible, but such assays can be technically challenging. In some cases, it may be possible to design manufacturing procedures that reduce the risk of problematic recombination events, and this is preferable to relying solely on testing.
Replication-competent adenovirus (RCA) may occur when E1-deleted adenovirus vectors are manufactured on complementing cell lines that contain adenovirus E1 sequences. Cell lines such as 293 cells contain sufficient adenovirus sequences that homologous recombination can occur, thus introducing functional E1 sequences into an E1-deleted vector. FDA recommends that there be less than one infectious RCA in 3×10 10 vector particles . The risk of RCA formation can often be reduced by changing the manufacturing conditions. One strategy is to use more modern E1-containing cell lines that have little or no homology to the vector sequences, thus greatly reducing the frequency of recombination. Another strategy to reduce the likelihood of RCA is to delete additional essential genes from the adenovirus vector so that multiple recombination events would be needed to create RCA.
Retroviral vectors and lentiviral vectors also present a risk of recombination events that may result in replication-competent retroviruses (RCR or RCL). If cell therapies are cultured for a significant amount of time after transduction with retroviral or lentiviral vectors, FDA also recommends that these cellular products be tested for the presence of replication-competent viruses . Although time-consuming and labor-intensive, the most sensitive tests for RCR and RCL are based on amplification of potential recombinants by in vitro culture followed by an endpoint assay for detection. As with adenoviruses, however, improvements in vector design and manufacturing processes are helping to make retroviral and lentiviral vectors less susceptible to formation of RCR and RCL, and sponsors are well advised to design their manufacturing process to minimize the chances of unwanted recombination.
Testing for Contamination by Adventitious Agents
The culture-based methods that are used to manufacture biologics are inherently susceptible to contamination with adventitious agents. Adventitious agents can be detected using culture-based assays ( in vitro assay) or in animals ( in vivo assay). However, it may be particularly difficult to test certain virus-based cancer gene therapies for contamination by adventitious viruses because the product may replicate in the cells or animals that are used for these tests. Even with replication-incompetent viral vectors (e.g., adenovirus), the amount of test article must be carefully controlled to ensure that it is not toxic for cells in the in vitro assay. In many cases, neutralizing the product with a specific antiserum can reduce toxicity, but if this is insufficient, it may be necessary to consider alternative assays for adventitious viruses.
Especially in cases in which it is difficult to test for adventitious viruses, it is important to design manufacturing processes that minimize risks of contamination. In addition to rigorously following cGMPs, methods for reducing risk of contamination include avoiding animal-derived materials, use of disposable bioreactors and materials, and use of purification processes that may remove or inactivate undesirable infectious agents.
In summary, early attention to CMC can have a beneficial impact on the later course of product development. Although more work may be required at the start of a development program, attention to these issues can save time and money in the long term and will ease the eventual transition to late-phase trials.
Pharmacology/Toxicology Review
The administration of a gene therapy product into subjects in a clinical trial will first entail the conduct of preclinical studies in order to provide a pharmacology and toxicology profile that can help define the risk/benefit ratio of the investigational product in the target clinical population. In addition to providing adequate safety support for the initiation of a clinical trial, preclinical studies are also performed throughout the product development program, as appropriate.
The Initial Step: Proof-of-Concept Studies
Various approaches using gene therapy products for cancer treatment have been attempted. Examples include (1) replacement of a missing or altered gene (e.g., tumor-suppressor p53) ; (2) expression of transgenes to enhance tumor killing or sensitize cancer cells to chemo/radiotherapy (e.g., mda-7 or BikDD) or to alter the tumor microenvironment (e.g., bacteria-mediated delivery of genes with anti-angiogenesis effects) ; (3) expression of an enzyme that activates an anticancer prodrug ; (4) expression of silencing oligonucleotides (e.g., short hairpin RNA (shRNA)) that interfere with specific molecules responsible for tumor development ; (5) gene-modified cells as immunotherapies ; and (6) oncolytic viruses with or without a transgene against specific tumor types . For such diverse therapeutic approaches, proof-of-concept (POC) studies help to provide the supportive scientific justification and rationale for the use of a particular product in a specific disease indication. Important goals of POC studies are to provide insight into the product’s mechanism of action and to characterize the bioactivity profile involving the cancer cell target(s).
The in vivo POC studies should help establish the basis for conducting the desired clinical trial through the generation of data to show (1) a pharmacologically active dose range, with establishment of optimal biological dose (OBD) and minimally effective dose (MED) levels, and 2) a potentially optimal route and dosing schedule for product administration. State-of-the-art science and accompanying technologies should be incorporated into POC studies, as feasible, and identification of potential cancer biomarkers is also encouraged because they may assist in the overall clinical monitoring program.
Whenever possible, the gene therapy product that is used in the definitive POC and safety studies should be identical to the intended clinical product. For certain products, such as a gene-modified human cell that will likely be rejected in animals or a vector expressing a species-specific transgene, use of analogous animal cells or an animal analog gene can be considered.
Animal Models
Mice are a common species used in POC studies. Historically, the orthotopic tumor (xenograft/syngeneic) model is usually desired over the ectopic model because the orthotopic model is considered to better mimic the interaction of tumor cells with the local tissue environment . Frese and Tuveson suggest that genetically engineered mice (GEM), including transgenic and “humanized” mice, are more biologically relevant because many of these models better exhibit the pathophysiological and molecular features of human malignancies . Public resources, such as the NCI Mouse Models of Human Cancers Consortium, provide the cancer research community with various databases of murine cancer models including GEM . The use of currently available and appropriate tumor models is an important step to further understand and establish the pharmacological profile for a gene therapy product, which can provide guidance for clinical trial design.
In Vitro Studies
In vitro studies serve to supplement the in vivo animal data. These studies use human cells to provide additional insight into the biological activity of the gene therapy product, and they are an essential POC component, notably when in vivo testing in an animal model cannot be performed.
Safety Assessment
Preclinical safety assessment is a critical step toward administration of an investigational product in humans. For gene therapy products, this evaluation consists primarily of toxicology and vector biodistribution/persistence studies. These studies, along with POC studies, help formulate an acceptable clinical trial protocol. The overall principles of safety assessment that are used for the more established biotechnology-derived products (e.g., monoclonal antibodies, cytokines, and growth factors), as described in the International Conference on Harmonisation guidelines Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals (S6) and Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals S6(R1) , can apply to gene therapy products. However, the specific study designs to evaluate the safety of gene therapy products are based on a product-specific and (as applicable) a disease-specific approach.
Some gene therapy products are designed to induce specific antitumor immunity directly or by using a component of the immune system to target specific cancers. The general principles of preclinical study conduct for these products are included in the FDA Guidance for Industry: Clinical Considerations for Therapeutic Cancer Vaccines .
Animal Species/Models
If feasible, an existing animal model of disease is used in the safety studies to allow for assessment of the contribution of disease-related pathology to any toxicity observed. However, for products intended to treat cancer, the use of healthy biologically relevant animal species is often a reasonable choice. Usually, the majority of animal tumor-bearing models have a short life span, which may limit their practical use for long-term safety evaluation, as well as their ability to detect any potential adverse immune response to the administered gene therapy product.
Considerations in choosing a relevant animal species include (1) comparability to relevant aspects of human physiology; (2) biological response to the expressed transgene; and (3) biological response to the vector, virus, or cell population (if applicable).
For safety assessment of a replication-competent oncolytic virus, both virus infectivity and the pathological effect of viral infection are important considerations when selecting appropriate animal species. For example, Deisboeck et al . provided data showing that in addition to the historical use of the Aotus monkey, the common marmoset could also serve as a relevant species to assess the potential for neurotoxicity as a result of administration of herpes simplex virus-1 (HSV-1) . Similarly, Sonabend et al . report that Syrian hamsters and cotton rats are permissive to an oncolytic adenovirus with signs of hepatic toxicity and immune response to the virus . Another important aspect when selecting appropriate animal species for safety evaluation of a gene therapy is the ability of the species to biologically respond to an expressed human transgene. If the expressed human transgene is biologically active only in nonhuman primates, the sponsor should provide justification for the use of another species. Appropriate scientific justification should accompany the selection of the relevant animal species/models for use in the safety assessment of the gene therapy product. In some cases, with adequate justification, use of the clinical vector expressing an analogous animal transgene is an option.
Design of Toxicology Studies
The safety assessment of a gene therapy product should be comprehensive enough to identify, characterize, and quantify any potential local and systemic toxicity observed. Outcomes measured should include acute/chronic effects, resolution of any findings observed, delayed toxicity, and any dose–response effects . There are also broad safety concerns that may be related to immunogenicity of a specific vector/virus type or transgene and the delivery procedure. The study design may need to be individualized for a gene therapy product to address specific safety concerns, such as the replication-competent nature of oncolytic viruses; the potential on- and off-target toxicities following expression of silencing oligonucleotides such as shRNA; and the potential for oncogenesis by integrating vectors, such as retroviruses . The data generated from the toxicology studies for a gene therapy product should allow a determination of a dose level that is the highest dose level at which no biologically or statistically significant increase in the severity or frequency of adverse findings in morphology, function, or other observation parameters (e.g., mild serum chemistry or hematology changes of no clinical significance) compared to appropriate concurrent controls, is observed. This dose level is termed a “no-observed-adverse-effect level” (NOAEL). The extrapolation of this NOAEL to the clinical dose level range is accomplished by using scaling factors, such as body weight, body surface area, or organ weight/volume.
Vector Biodistribution
Biodistribution (BD) analysis is performed to determine the distribution and the persistence of the vector/virus to target and nontarget tissues following direct in vivo administration in animals. This analysis is not usually conducted on ex vivo transduced cells. This assessment is an important part of the overall safety profile of the gene therapy product and should guide the toxicology study design (i.e., possible target tissues, sacrifice intervals, and study duration). BD studies should be performed prior to product administration in humans if (1) the gene therapy product belongs to a novel vector or virus class, (2) there is little or no published safety data on the vector/virus or transgene of interest, (3) a significant modification or manufacturing change has been made to a vector/virus already in clinical trials, (4) the route of administration or the dosing schedule is changed, or (5) the existing clinical formulation is significantly changed.
BD analysis is generally conducted at the molecular level using a methodology that provides high sensitivity. The current “gold standard” for BD studies is a quantitative polymerase chain reaction (Q-PCR) assay that detects vector/virus genomes in biological fluids and tissue samples. For details regarding the Q-PCR assay, as well as recommendations on general study design and tissue collection procedures, refer to the FDA Guidance for Industry: Gene Therapy Clinical Trials—Observing Subjects for Delayed Adverse Events .
Other methodologies, such as nonterminal imaging techniques, may also offer a potential modality for monitoring the BD of a gene therapy product in both animals and humans. Räty et al . published a review of several methods of noninvasive imaging of GT products, which included magnetic resonance imaging, single-photon emission tomography/positron emission tomography, and fluorescence and bioluminescence imaging .
Good Laboratory Practice
Preclinical studies performed to determine the safety of a test article (i.e., the investigational gene therapy product) should be conducted in compliance with good laboratory practice (GLP), in accordance with the U.S. regulation 21 CFR Part 58. Preclinical in vitro and in vivo studies conducted to support POC do not need to be conducted under GLP. However, it is also understood that in some instances, such as the nature of the animal model used or the need to perform specialized assays, the safety studies may not be in compliance with GLP. These studies can still be acceptable as long as they are performed in accordance with a prospectively designed protocol and the data are of sufficient quality and integrity to support the proposed clinical trial. The complete study report that is submitted to FDA/CBER should include a section that identifies any areas that deviate from the protocol and the potential impact of these deviations on study integrity.
Summary
The preclinical program is an important aspect for enabling transition from the lab to the subject. This section has touched upon some of the basic concepts of preclinical study design for gene therapy products. Another resource for a comprehensive summary regarding preclinical considerations for gene therapy products is the OCTGT Learn Webinar Series . A well-designed preclinical program is essential to establishing an acceptable risk/benefit profile for each investigational gene therapy product, enabling evolution from translational research to clinical trial. For a comprehensive summary regarding preclinical assessment of gene therapy products, see the document titled Draft Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products (November 2012) at https://www.federalregister.gov/articles/2012/11/29/2012-28882/draft-guidance-for-industry-preclinical-assessment-of-investigational-cellular-and-gene-therapy .
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Clinical Trials Using LV-P140K-MGMT for Gliomas
 Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


