Fig. 10.1
A 3-month old with Beckwith–Wiedemann Syndrome who was born with hyperinsulinism and an omphalocele who later presented with a left adrenal mass. a Macroglossia and hirsutism (forehead). b Gynecomastia (black arrow) and a repaired omphalocele (white arrow). c Hirsutism of the leg

Fig. 10.2
a CT Scan demonstrated a left adrenal mass consistent with an ACT (white arrows). b An open left adrenalectomy was performed which confirmed an ACT with no lymphatic or vascular invasion and all nodes negative of disease
ACTs have a high frequency of chromosomal gains and amplifications, and several chromosomal subregions containing candidate proto-oncogenes have been identified [13, 14]. The most consistent findings in ACTs have been the presence of copy number gains in chromosome region 9q34 [15]. However, there is no consistent difference in chromosomal gains or losses between the adrenal adenomas and carcinomas [15].
In rare cases, ACTs have been reported in association with congenital urinary tract abnormalities such as duplication of the collecting system, with tumors such as ganglioneuroma and ganglioneuroblastoma, and with congenital adrenal hyperplasia [16–20]. Environmental exposures to several agents have been postulated to be important in the development of ACTs, but these have typically been case reports with unclear mechanisms [21, 22].
Epidemiology of Adrenocortical Tumors in Children
Of the adrenal tumors of childhood, ACTs are less common than neuroblastomas but are more common than pheochromocytomas [23]. Neuroblastomas and pheochromocytomas derive from the adrenal medulla while ACTs are the most common tumor of the adrenal cortex in childhood [24, 25]. Even so, ACTs are rare in American children. It is estimated that there will be about 19 new cases of adrenocortical carcinomas in children per year in the US [26]. However, some cases of adrenocortical carcinoma may be initially misclassified as adrenocortical adenoma, so a better estimate is probably 25–30 cases per year [27]. A recent Surveillance, Epidemiology, and End Results (SEER) program study review of pediatric adrenocortical carcinoma calculated an incidence for patients under 20 years of age at 0.21 per million [28].
Interestingly, there are geographic differences in the incidence of ACT. In a region of southern Brazil, ACTs are much more common. The markedly increased incidence of ACTs in this area is probably due to the increased incidence of a p53 mutation in the population although other genetic factors and environmental exposures have been suggested.
ACTs typically present in the first five years of life with a median age of presentation between 3 and 4 years of age. However, there is a biphasic age distribution with a larger peak during infancy and a second smaller peak in adolescence [29–32]. Girls outnumber boys in all reports, with a female to male ratio of 1.5:1. This female predominance increases after adolescence to 6:1 [29, 31, 32].
Pathology of Adrenocortical Tumors in Children
Unlike similar tumors in adults, adrenocortical adenomas in children have no histopathologic features that allow them to be reliably distinguished from adrenocortical carcinomas. Also, the biologic behavior of pediatric adrenocortical neoplasms may be difficult to predict on the basis of morphologic criteria. Thus, the term adrenocortical neoplasm [or adrenocortical tumor (ACT)] is currently used to designate both benign and malignant tumors of the adrenal cortex in children [10].
Gross Features of Adrenocortical Tumors in Children
The median tumor weight of an ACT is 126 g but may range from 2 g to 6 kg. There is no tumor laterality predominance, and bilateral tumors occur in about 1% of cases [33, 34]. There are descriptions of ectopic sites within the spinal canal [35], thoracic cavity [36] and in the abdomen away from the adrenal gland. Ectopic occurrence is not surprising in view of the adrenal’s close proximity to the celiac plexus, kidney, genitalia, broad ligament, epididymis, and spermatic cord during development.
Adenomas are usually spherical, unilateral, solitary, and well demarcated, but often not truly encapsulated. They typically weigh less than 50 g. Adenomas range in color from yellow to red-brown, but may appear black if the tumor contains a large amount of the pigment lipofuscin [37, 38].
Carcinomas usually weigh over 100 g although smaller malignant tumors have been reported. Grossly, they have coarse trabeculations, a multinodular contour, and are yellow to brown in color. Areas of hemorrhage and necrosis are frequently seen [38, 39]. In adrenocortical neoplasms of childhood, malignant behavior is usually associated with lesions that weigh more than 500 g, whereas most tumors that weigh less than 500 g are benign [40]. Cystic changes may be seen in both adenomas and carcinomas, but are more common in carcinomas and larger adenomas [10].
Microscopic Features of Adrenocortical Tumors in Children
Adrenal adenoma comprises a heterogeneous group of benign neoplasms that histologically resemble the appearance of the normal zona fasciculata, the zona glomerulosa, or most often, a combination of both [10]. Cells are typically arranged in nests separated by a delicate fibrovascular stroma. Cytological features vary from large, pale vacuolated cells with vesicular nuclei characteristic of the zona fasciculata to smaller cells with eosinophilic cytoplasm and condensed chromatin similar to those in the zona glomerulosa and zona reticularis [10]. In general, adrenocortical adenomas are histologically bland, with low nuclear-to-cytoplasmic ratio, very little necrosis or hemorrhage, and they rarely have mitoses or bizarre nuclear forms. In children, however, benign adrenocortical tumors are more likely to display marked nuclear atypia, polymorphism, necrosis, and mitotic activity than do similar tumors in adults. Occasionally, central degenerative changes (necrosis, hemorrhage, cystic alterations, and vascular proliferation) may be seen in adenomas. Despite some association of cell types with secretory products, it is not possible to reliably predict the endocrine function of a tumor on the basis of histologic characteristics [10].
Adrenocortical carcinoma shows a wide range of differentiation on histologic examination, not only between different tumors but also within the same tumor [10]. Morphology ranges from normal-appearing adrenal cells to completely undifferentiated cells. Broad fibrous bands often separate the tumor into multiple nodules. Most cells are lipid-poor and eosinophilic, and they may be arranged in nests, trabeculae, or sheets. Hyperchromatic nuclei, pleomorphism with bizarre giant cells and multinucleated forms, necrosis (especially confluent areas of necrosis), mitotic activity (including atypical mitoses), and vascular or capsular invasion may be seen. However, none of these histologic features is necessarily diagnostic of malignancy in ACTs in children [6, 26].
Adrenocortical Adenoma or Carcinoma?
There have been several attempts to distinguish between adrenocortical adenomas and carcinomas using clinical and pathologic features of the tumors. These studies have analyzed many different parameters with varying degrees of success. In children, clinical findings, tumor size, tumor weight, and histologic features may suggest malignant potential, but no single parameter (except the detection of metastases) allows benign tumors to be discriminated from malignant ones [10, 41]. Classification of carcinomas into low grade and high grade based on mitotic rate has also been proposed [42, 43]. However, ACTs in children cannot be reliably classified as benign or malignant using these systems [42, 44].
Special studies are often of limited value in the pathologic diagnosis of ACTs. Adrenocortical carcinomas are usually vimentin-positive and often negative for cytokeratin, epithelial membrane antigen, and carcinoembryonic antigen [10]. However the main value of immunohistochemistry of an adrenal mass is to diagnose neoplasms that metastasize to the adrenal gland rather than distinguishing between benign and malignant ACTs [45].
The electron microscopic appearance of adenomas resembles that of cells of the normal adrenal cortex, with steroid-producing cells showing tubulovesicular or tubular lamellar mitochondria, and an abundant, smooth endoplasmic reticulum. Adrenocortical, carcinomas may show abnormal numbers and morphology of mitochondria, and dissolution of the basement membrane surrounding alveolar groups of cells but these findings are not diagnostic [26].
Chromosomal analyses of ACTs has shown that aneuploidy tends to point toward malignancy, although this characteristic is also found in benign tumors, especially larger ones [46, 47]. Cytogenetic studies of carcinomas have shown loss of heterozygosity of several gene loci in some cases [47, 48]. Mitotic rate has been consistently reported as the most important determinant of aggressive behavior [43, 49–51]. Other histopathologic variables are also important, and it is possible to stratify the risk of recurrence based on a score that includes a combination of different histopathologic characteristics, such as venous, capsular, or adjacent organ invasion, tumor necrosis, mitotic rate, and the presence of atypical mitoses [51].
An unanswered question is how to categorize an ACT with a few “malignant” pathologic features. One approach proposed by Bugg [42] is to differentiate between those adrenocortical carcinomas with low-grade pathologic features and those with high-grade pathologic features. Another approach advocated by Dehner is to acknowledge the uncertainty by using terms such as “atypical adrenocortical neoplasm” or “adrenocortical neoplasm of uncertain or indeterminate malignant potential” for the completely resected tumor and recommend close follow-up with appropriate hormonal monitoring [52].
Evaluation of Adrenal Masses
The combination of history, physical examination, laboratory tests, and diagnostic imaging can usually distinguish adrenal cortical tumors from adrenal medullary tumors and other adrenal problems. Modern cross-sectional imaging in particular is critical in the evaluation of children with clinical features of adrenocortical hyperfunction. Finding an adrenal mass in this setting is diagnostic of an adrenocortical neoplasm.
Symptoms and Signs of Adrenal Masses
Adrenal tumors usually present with mass effects or with systemic manifestations of excessive hormone secretion. A neuroblastoma can grow quite large and present as a palpable abdominal mass or with symptoms of compression of surrounding structures, such as when the tumor extends into the spinal canal and causes paralysis. Neuroblastoma may also present with signs of metastases such as periorbital lesions causing bulging eyes or dark circles around the eyes (“black eyes” or “raccoon’s eyes”), bone metastases causing bone pain, liver metastases leading to hepatomegaly and respiratory embarrassment, and subcutaneous deposits resulting in bluish lesions under the skin (“blueberry muffin”). Finally, both neuroblastoma and more commonly pheochromocytoma can present with systemic effects of catecholamine secretion such as hypertension, palpitations, sweatiness, shakiness, anxiety, and diarrhea.
In contrast to adult ACTs, most pediatric ACTs secrete hormones that lead to their presentation, usually at 5–8 months of age [29, 32, 53]. Children will present with various manifestations depending on the specific adrenocortical hormones secreted by the tumor. The most common presentations are virilization, feminization, Cushing’s syndrome, and Conn’s syndrome. Although the clinical manifestations of one endocrine syndrome may predominate, ACTs usually secretes several hormones and thus signs and symptoms of multiple syndromes may be seen.
Precocious puberty refers to secondary sex characteristics that appear in girls younger than 8 years, and in boys younger than age 9. Precocious puberty may be gonadotropin-dependent (true precocious puberty) or gonadotropin-independent (pseudoprecocious puberty). Furthermore, precocious puberty may be characterized as isosexual when the premature secondary sex characteristics are appropriate for the patient’s gender, or heterosexual when the premature secondary sex characteristics are inappropriate for the patient’s gender. Heterosexual precocious puberty manifests as virilization in girls and feminization in boys. Because functioning adrenocortical neoplasms represent a gonadotropin-independent source of endogenous androgens and cortisol, they usually produce pseudoprecocious puberty, Cushing’s syndrome, or a mixture of the two [10].
Virilizing syndrome is the most common presentation, with about 80% due to the secretion of androgens, which cause deepening of the voice, acne, hirsutism, increased muscle mass, and secretion and proliferation of the sebaceous glands. Specifically, in girls, there is clitoral enlargement, facial and pubic hair, amenorrhea, advanced bone age, and occasionally male pattern hair changes [10]. In boys, there is early development of acne, pubic hair, penile enlargement, and precocious isosexual pseudopuberty [10].
Cushing’s syndrome results from the production of autogenous corticosteroids and occurs in about one third of patients with ACTs. Children with Cushing’s syndrome present with moon facies, weight gain, centripetal distribution of fat, plethora, hypertension, and striae. In addition, most patients who present with Cushing’s syndrome will also have signs and symptoms of virilization.
Conn’s syndrome is also known as primary aldosteronism and may be caused by adrenal cortical carcinoma, adrenal adenoma, or bilateral cortical hyperplasia. Aldosterone-producing adenoma is rare in children [54]. Symptoms associated with Conn’s syndrome include headache, weakness of proximal muscle groups, polyuria, tachycardia, hypocalcemia, and hypertension. As with Cushing’s syndrome, a patient’s presentation may be complicated by manifestations of other hormones secreted by the ACT.
Laboratory Investigation of Adrenal Masses
The laboratory investigation of adrenal masses includes measuring the abnormal hormone secretions of medulla (catecholamines) or cortex (mineralocorticoids, glucocorticoids, androgens) and evaluating systemic effects of the tumor or its abnormal hormonal production. When medullary tumors are suspected urine or blood may be collected for substances secreted by the tumor including homovanillic acid (HMA), vanillylmandelic acid (VMA), dopamine, and norepinephrine.
Laboratory testing for ACTs includes evaluation of androgen and glucocorticoid levels and their breakdown products. Measurements of urinary 17-ketosteroids (17-KS) are the most important marker for ACT. 17-KS levels are elevated in the majority of patients with ACTs regardless of whether the tumor is associated with Cushing’s syndrome or virilization [31]. Plasma dehydroepiandrosterone sulfate (DHEA-S) levels are abnormal in about 90% of patients with ACTs, making this the second most sensitive tumor marker. Abnormal urinary DHEA is less sensitive, with only two thirds of cases with detectable elevation. Urinary 17-hydroxycorticosteroid (17-OH) levels are elevated in patients with clinical signs of excessive glucocorticoids. Therefore, evaluation of patients suspected of having an ACT includes determination of urinary 17-KS and 17-OH, urinary or plasma cortisol (either baseline or suppressed), plasma DHEA-S, testosterone, androstenedione, 17-hydroxyprogesterone, aldosterone, renin activity, 11-deoxycorticosterone (DOC) and other 17-deoxysteroid precursors. The hormone activity will not help differentiate benign from malignant lesions since both will have a significant elevation in these markers [55]. Additional useful tests include serum electrolytes, especially potassium, which would be low with increased aldosterone, and serum glucose, which can be elevated with, increased corticosteroids.
Imaging Evaluation of Adrenal Masses
By the time that most children present with an adrenal mass, the mass is visible with diagnostic imaging; however, small lesions with hormonal activity may be radiologically occult. With current imaging technology, the scenario of delayed diagnosis of a functional adrenal tumor that was common in the past has largely been eliminated [23, 25].
Plain radiography has limited value but can reveal a mass effect of the tumor on surrounding organs or calcifications, which suggest neuroblastoma or previous adrenal hemorrhage. In the past excretory urography and nephrotomograms were often performed, which would show the mass effect of an adrenal lesion on the kidney and urinary collecting system however, these tests have been replaced by ultrasound and cross-sectional imaging.
Ultrasonography (US) is often the first imaging procedure performed in children with abdominal masses since it can be performed easily and without sedation. US can determine the location of a mass and its relationship to surrounding vessels and organs, determine if the mass is cystic or solid, assess the vascularity of the mass, and identify associated liver metastases. US serves as a good initial study to define the next step of management (Fig. 10.3a). However, US is most useful for large masses and the majority of lesions of the adrenal cortex tend to be small and are often difficult to visualize. Doppler US is a valuable tool to evaluate for associated tumor extension into the renal vein, inferior vena cava, and right atrium (Fig. 10.3b). Hamper et al described the sonographic features of ACTs including a wide range in size (range 3–22 cm), a round or ovoid circumscribed shape commonly displaying a lobulated border, and in a quarter of the cases, a thin echogenic capsule-like rim. The adrenocortical carcinomas less than 6 cm were homogenous solid masses and were nearly isoechoic to renal cortex. Larger lesions tended to be heterogeneous and to contain central or diffuse hypoechoic regions that corresponded to necrotic areas in the surgical pathology specimens. The term “scar sign” refers to radiating linear echoes that suggest adrenocortical carcinoma [56].
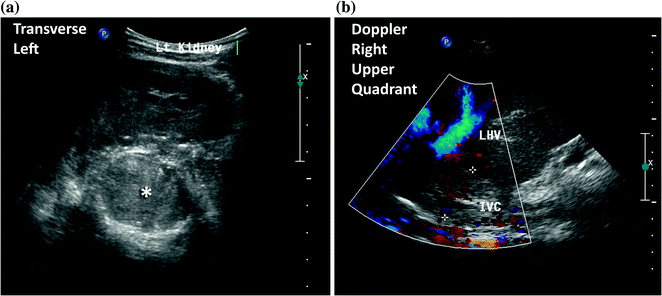
Fig. 10.3
An 11-year-old female presented with a large left sided abdominal mass. A CT scan and MRI were performed which demonstrated a left adrenal primary tumor with tumor extension through the adrenal veins into bilateral renal veins, hepatic veins, IVC, and into the right atrium. Her treatments consisted of biopsy, neoadjuvant chemotherapy, resection of primary and thrombus under cardiopulmonary bypass, and Mitotane therapy. a Transverse ultrasound of the left upper quadrant demonstrating the left kidney displaced inferiorly and a large mass in the left suprarenal space (*). b Doppler ultrasound of the right upper quadrant demonstrating a partial occluding tumor thrombus (+) in the inferior vena cava (IVC)
Computerized Axial Tomography (CT) is the imaging procedure of choice for evaluating adrenal lesions. CT provides accurate definition of the adrenal size, location, and appearance (Fig. 10.4a). Furthermore, it assesses local and vascular invasion, enlargement of lymph nodes, and the presence of metastases within in the abdomen or distant sites (Fig. 10.4b). On CT, ACTs are typically circumscribed, appear variably heterogeneous due to hemorrhage and necrosis, and may display a thin capsule-like rim [10]. Larger lesions tend to enhance heterogeneously with IV contrast.
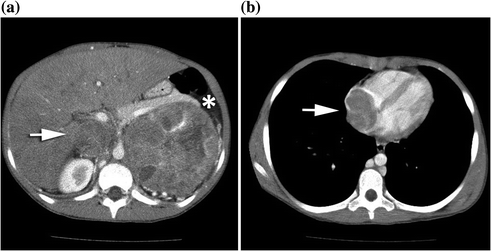
Fig. 10.4
An 11-year-old female presented with a large left sided abdominal mass. a Axial CT scan with a 10.1 X 10.2 cm heterogeneous left adrenocortical carcinoma (*) displacing the pancreatic tail superiorly and the left kidney inferiorly with left renal vein and IVC tumor thrombus (white arrow). b Axial CT scan with more cranial views demonstrating right atrial tumor thrombus (white arrow)
Magnetic resonance imaging (MRI) may also define the extent of the adrenal lesion including its relationship to surrounding organs and vessels (Fig. 10.5). It may provide some advantages over CT, as it involves no radiation, displays different planes, and may better characterize tumor thrombus (Fig. 10.5). ACTs have intermediate signal intensity on T1-weighted MR images, and high signal intensity relative to liver on T2-weighted images [10]. Also, MR may help differentiate benign from malignant lesions based on the enhancement with gadolinium [57].
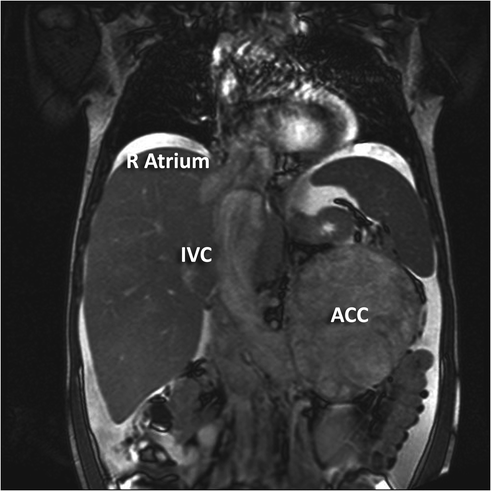
Fig. 10.5
An 11-year-old female presented with a large left sided abdominal mass. Coronal MRI demonstrating left adrenocortical carcinoma (ACC) with left renal vein and IVC tumor thrombus (IVC) and extending into the right atrium (R Atrium)
Nuclear scans may also be helpful in the evaluation of adrenal masses in some specific situations. Metaiodobenzylguanidine (MIBG) scans may be used to detect adrenal medullary tumors, pheochromocytomas, and neuroblastomas due to MIBG affinity for medullary tissue. MIBG scans may be particularly useful in localizing such tumors in extra-adrenal sites. Iodocholesterol-labeled analogs may be used to detect primary adrenocortical tumors or metastases. Dexamethasone administered before the scan may make the test more sensitive by suppressing autologous ACTH-responsive adrenal tissue.
More recently, positron emission technology (PET) scanning has been used to study recurrent or metastatic adrenal tumors. However, its role has yet to be fully elucidated.
Differential Diagnosis of Adrenal Masses
The differential diagnosis of an ACT includes a variety of benign and malignant conditions and some of the more common considerations are discussed below.
Neuroblastoma
Neuroblastoma is a neoplasm of the adrenal medulla or extra-adrenal sympathetic tissue that typically affects young children. The age at diagnosis and some clinical manifestations may overlap with the presentation of ACTs, however, neuroblastomas usually produce excessive catecholamines that can be detected in the serum and urine. Also, children with neuroblastoma are much more likely to present with metastases compared to children with ACTs. Furthermore, neuroblastomas may be associated with paraneoplastic syndromes of myoclonic encephalopathy and tumor elaboration of vasoactive intestinal peptide.
On imaging the typical neuroblastoma is a large, irregular, retroperitoneal mass that frequently encases vascular structures, often contains characteristic punctate calcifications, and may extend through neural foramina into the extradural spinal canal. However, neuroblastoma may also appear as a circumscribed suprarenal mass that is indistinguishable from an ACT. One imaging study that may be helpful in distinguishing an ACT from a neuroblastoma is the MIBG scan, which will be positive in patients with neuroblastoma.
Pheochromocytoma
Pheochromocytoma is a neoplasm derived from neural crest tissue. Similar to neuroblastoma, a pheochromocytoma secretes catecholamines and concentrates MIBG, however, pheochromocytomas usually occur in older children (6–14 years) compared to neuroblastomas and ACTs that usually occur in infants and toddlers. Children with pheochromocytomas typically present with constant or paroxysmal hypertension with resulting headaches [58, 59]. Pheochromocytomas may occur in patients with neurofibromatosis, von Hippel-Lindau disease, Sturge-Weber syndrome, and multiple endocrine neoplasia types IIA and IIB. Less than 10% of pheochromocytomas in children are malignant [10]. On imaging, they tend to be rounded, circumscribed masses. On T1-weighted MR, a pheochromocytoma has lower signal intensity than that of the liver and on T2-weighted images a higher signal intensity than ACTs [59].
Adrenal Hemorrhage
Adrenal hemorrhage typically occurs in neonates, which would be an unusual age of presentation for adrenocortical tumors. Adrenal hemorrhage can be distinguished from a neoplastic adrenal mass by serial sonograms that demonstrate a temporal evolution of the mass through stages of liquefaction, clot retraction, and eventual shrinkage.
Renal Lesions
Large adrenocortical neoplasms may appear on imaging to invade or arise from the upper pole of the kidney. Solid renal neoplasms of childhood include Wilms tumor (nephroblastoma), mesoblastic nephroma, renal cell carcinoma, clear cell sarcoma, and rhabdoid tumor of the kidney. None of the renal tumors produces the clinical findings of hormonally active ACTs. However, rhabdoid tumor and mesoblastic nephroma may be associated with hypercalcemia.
ACTH-Independent Macronodular Adrenal Hyperplasia (AIMAH)
AIMAH, also known as massive macronodular adrenocortical disease (MMAD) [60–62], is a benign proliferative disorder of the adrenal cortex that presents with ACTH-independent Cushing’s syndrome. Steroid hormone secretion is ACTH independent and associated with both undetectable plasma levels of ACTH and the inability to suppress cortisol secretion with high dose dexamethasone [63]. Histologically, it is composed of nodules with two cell types, lipid rich cells with clear cytoplasm and lipid-poor cells with a compact cytoplasm [60]. Although corticosteroid secretion is independent of ACTH the cells express the ACTH receptor and patients will respond to exogenous ACTH [61, 64]. The increased steroid hormone synthesis in AIMAH is thought to be due to an overall increase in adrenocortical mass rather than augmented synthesis within each cell [64]. AIMAH may be associated with McCune-Albright syndrome, an autosomal dominant disorder characterized by polyostotic fibrous dysplasia, café-au-lait spots, precocious puberty, and hyperfunctioning endocrine glands [65]. McCune-Albright syndrome is caused by an activating mutation in the GNAS1 gene.
Primary Pigmented Nodular Adrenocortical Disease (PPNAD)
PPNAD is similar to AIMAH but it does not respond to ACTH [61, 66]. Both AIMAH and PPNAD are benign proliferative disorders. PPNAD is characterized by nodules usually less than 4–6 mm in diameter with a brown or black color. The color is a result of large cells containing a granular, pigment-containing cytoplasm. The internodular adrenal cortex is atrophic and disorganized, and the adrenal glands are usually normal weight and size [67, 68]. PPNAD can be seen isolation, but it is usually associated with the Carney complex, an autosomal dominant syndrome that includes perioral, ocular, or genital schwannomas, and endocrine gland hyperactivity. Affected patients often have tumors of two endocrine glands. The most common tumor in the Carney complex is PPNAD and other tumors include prolactin or growth hormone secreting pituitary tumors, thyroid adenomas or carcinomas, testicular large-cell calcifying Sertoli cell tumors, and ovarian cysts [69–71]. Clinically evident PPNAD is seen in 25–30% of patients with Carney complex and it usually presents in childhood, late adolescence, or early adulthood [67, 71, 72]. Such patients have been treated successfully with bilateral adrenalectomies [68].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


