Fig. 14.1
The two-hit model of acute leukemogenesis. This model hypothesizes that AML and ALL are the consequence of a combination of at least two broad classes of mutations. Class I mutations involve an activating lesion in signaling pathways and confer a proliferative and/or survival advantage to hematopoietic cells. Class II mutations lead to an arrest of lymphoid or myeloid differentiation as a result of loss of function of transcription factors or cofactors that are important for normal hematopoietic differentiation
Cumulative evidence from different murine models of leukemia provide convincing support for the necessity of cooperative transforming events (Class I events) in addition to the expression of fusion genes (Class II events) for the development of acute leukemia. For example, expression of PML–RARA, the product of a gene fusion associated with a subtype of AML, under the control of the Cathepsin G promoter in transgenic mice, results in altered myeloid development, but PML–RARA alone is not sufficient to directly cause AML [2]. Similarly, expression of RUNX1–CBFA2T1, another common gene fusion in AML, does not produce acute leukemia in mice until combined with a dose of the chemical mutagenic agent, N-ethyl-N-nitrourea (ENU) [3]. This two-hit phenomenon repeats itself with the other common fusions associated with human leukemia [4, 5]. A study of monozygotic twins who both developed ETV6–RUNX1 positive ALL [6] provides further support for the requirement of secondary genetic hits. The specific genomic breakpoint in fusions is essentially unique to each patient with leukemia, however the twin leukemic DNA in this study shared an identical ETV6–RUNX1 fusion sequence. The most plausible explanation for this finding is that the chromosome translocation event occurred in utero. However, despite possessing identical ETV6–RUNX1 gene rearrangements in an identical genetic background from birth, the ALL manifested itself only later in life and with differential timing between the twins. Additionally, the concordance rate for ALL in syngeneic twins is estimated at only 5 % [7]. Together these data indicate that the intrauterine translocation event is insufficient for clinical leukemia and that a postnatal promotional event also occurs to induce leukemia [8]. Thus, both animal models and clinical observation support the notion that two cooperating genetic insults, leading to a proliferative advantage and to impaired hematopoietic differentiation, are necessary for the clinical manifestation of acute leukemia.
Common Etiological Issues
Leukemogenesis is a multistep process that requires the accumulation of alterations in a hematopoietic progenitor cell at multiple stages. The different subtypes of AML and ALL may have different causal mechanisms, as evidenced by their characteristic cytogenetic abnormalities. Considerable advances in the understanding of leukemogenesis have been made by studying the subtype-specific gene mutations and cytogenetic abnormalities, particularly reciprocal translocations. However, there are some particularly important concepts that are relevant in the pathogenesis of all acute leukemias and these will be considered in the next sections.
Leukemic Stem Cells
Hematopoietic stem cells (HSCs) are pluripotent stem cells that give rise to all the circulating blood cell types. They are defined by their ability to self-renew while generating differentiated daughter cells [9]. The maturation of HSCs into blood cells has been suggested to follow a hierarchical model (Fig. 14.2), in which the stem cells undergo a reduction in self-renewal potential while simultaneously gaining functional specialization [10]. Early on, HSCs make a fundamental commitment to either the lymphoid or myeloid fate. These two precursor types subsequently undergo further restriction in developmental potential until terminally differentiated cells are generated [11]. The common myeloid progenitors give rise to monocytes, neutrophils, basophils, eosinophils, erythrocytes, or megakaryocytes, while the common lymphoid progenitors give rise to B or T cells [12].
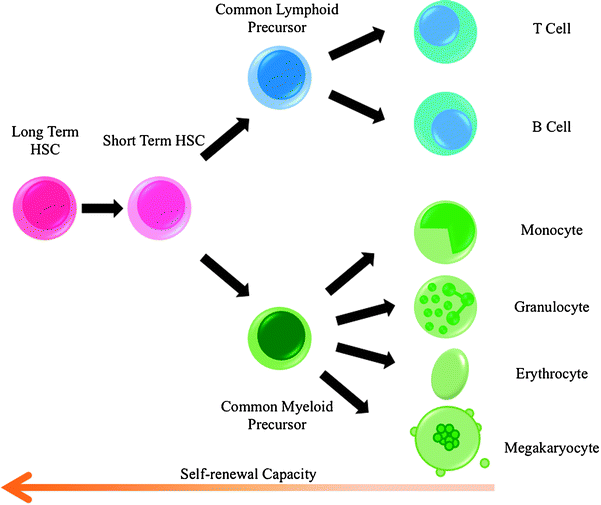
Fig. 14.2
The hierarchy of hematopoietic development. Long-term stem cells (HSCs) develop into different precursor cells whose self-renewal capacity and developmental potential become progressively more restricted
Cancer is a clonal disease that initiates in a single cell that has accumulated sufficient genetic damage to cause uncontrolled proliferation, and therefore the progeny of this cell makes up the tumor [13]. The experimental setting of transplantation, where subsets of tumor cells are transferred from one organism to another, demonstrates that tumors are functionally heterogeneous, as only a certain subpopulation of cells has the capacity to maintain or reinitiate the tumor. It is not always easy to define which cells within the population have this capability for self-renewal or tumor re-initiation.
Two general, but distinct models have been proposed to account for the heterogeneous proliferative potential of tumor cells (Fig. 14.3). The stochastic model proposes that all cells within the tumor are equivalent with respect to their potential to re-initiate the clone, however, any one cell would have a low probability of exhibiting this potential. In contrast, the hierarchical model predicts that re-initiation potential is a property of only a distinct subset of cells within the tumor, the so-called “cancer stem cell” [14].
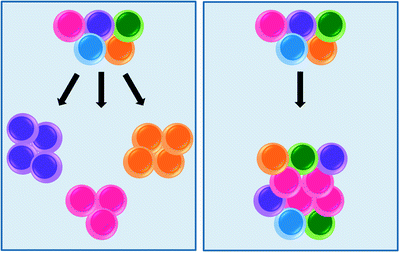
Fig. 14.3
Two models accounting for the heterogeneous proliferative potential of leukemic cells. The stochastic model (left panel) assumes that every cell in a tumor is potentially tumor initiating. Progression is governed by rare, random events. Cells with mutations that acquire growth advantage will dominate over all other cells in the tumor and will originate a new clone. In contrast, the hierarchical model (right panel) posits that acute leukemia consists of two functionally distinct cell types (1) leukemic stem cells (LSCs, pink), which are self-renewing cells with the capacity to initiate, sustain, and expand the disease, and (2) non-self renewing progeny cells, derived from LSCs through differentiation, which likely make up the bulk of the tumor and account for disease symptamaology
There is strong support for the idea that cancer, and in particular leukemia, is a stem cell disease. Persuasive evidence for this concept comes from the discovery that most AML blasts do not proliferate [15, 16] and that only 1–4 % of leukemic cells transplanted in vivo are able to form spleen colonies. The most compelling evidence for a leukemia stem cell (LSC), or leukemia initiating cell (LIC), has been provided by a Canadian group based at the University of Toronto. In a seminal paper in this field, Lapidot et al. [17] reported that xenograft transplantation of only a few, highly selected human LSCs into immunodeficient mice was able to recapitulate a leukemic disease that reproduced many features of the AML seen in the original human patients. Subsequently, cell purification based on cell surface markers demonstrated that the cells capable of initiating human AML in non-obese diabetic mice with severe combined immunodeficiency disease (NOD/SCID mice) were from a rare subpopulation of CD34+/CD38− cells. Normal HSCs share this same CD34+/CD38− phenotype, suggesting that normal primitive cells, rather than committed progenitor cells, are the target for malignant transformation.
Further studies support a multistep process whereby an initial molecular aberration occurs at the level of the stem cell, creating a pre-leukemic LSC. Either these cells or their progenitors then undergo additional oncogenic events that ultimately lead to transformation into overt disease [18, 19]. LSCs themselves also demonstrate self-renewal heterogeneity, with a population of quiescent stem cells capable of endless self-renewal, progenitor populations with more limited potential for self-renewal, and, finally, a population of blasts with no self-renewal potential that is responsible for the clinical manifestations of the disease [20]. This hierarchical arrangement of LSCs mirrors that of HSCs and lends credence to the idea that, in leukemia, the primary target for cell transformation resides within the HSC population.
The concept of the LSC has increasingly important implications for treatment of acute leukemia. Most conventional chemotherapies and even existing targeted therapies may fail to affect the LSC. This may be attributable to the fact that the majority of the LSC population exists in the G0 phase of the cell cycle, where the absence of DNA synthesis, cell division, or any activity related to self-renewal, renders it insensitive to anti-leukemic drugs. The induction of LSCs to enter the cell cycle, where they are then vulnerable to destruction by standard chemotherapy drugs, represents one therapeutic strategy. Indeed, Saito [21] and colleagues demonstrated in a mouse model of human AML that treatment with granulocyte colony-stimulating factor (G-CSF), a cytokine that induces cell cycle entry of hematopoietic stem cells, caused the transplanted leukemia stem cells to proliferate and rendered them susceptible to the chemotherapeutic cytarabine. This finding provides proof of principle that the eradication of leukemia at its roots can be accomplished by developing treatments focused on pushing LSCs out of their quiescent state. An analogous approach, in which newly diagnosed AML patients received chemotherapy, with or without G-CSF, showed the addition of G-CSF concurrently with chemotherapy improved disease-free survival [22].
Additionally, HSCs interact with their microenvironment, termed the “stem cell niche,” through interactions between the chemokine SDF-1 and its receptor CXCR-4, and by the adhesion molecules VCAM/VLA4 and Angiopoietin-1/TIE-2. The same molecules are also involved in leukemia stem cell–niche interactions, and elevated levels of CXCR4 and VLA4 have been associated with poor response to chemotherapy and unfavorable prognosis in AML. Therefore, it may be possible to affect leukemia stem cells directly by targeting the specific molecules mediating LSC–niche interactions. Finally, in addition to nonspecific treatments, such as G-CSF, that manipulate LSC behavior, targeting LSC-specific molecules may represent an effective therapy for AML. For example, specific targeting of the PML–RARA leukemic oncoprotein expressed in LSCs with arsenic trioxide may be the mechanism by which this treatment achieves long-term remission of murine, and possibly human, leukemia [23].
Although not all malignancies fit into the cancer stem cell model, AML in particular is one of the diseases for which there is solid evidence of stem cell-like behavior. Consequently, there is considerable leukemia research devoted to further understanding the unique biologic and molecular properties of LSCs, by comparison with both their non-self-renewing downstream progeny and their normal HSC counterparts.
Epigenetics
Epigenetic regulation refers to those heritable changes in gene function that occur without alteration of the primary DNA sequence [24]. The remodeling of chromatin, through DNA methylation and/or posttranslational modification of nucleosomal histones, has been implicated in the regulation of transcription. Anomalous epigenetic changes occur frequently in acute leukemia. The recurring chromosomal translocations seen in AML and ALL result in the generation of chimeric fusion proteins, as discussed in more detail later. Several of these fusion oncoproteins contribute to the development of leukemia partly by disrupting the modification of chromatin, through recruitment of chromatin-modifying coregulators [25]. The emerging link between the enzymes that modify chromatin and the transcription factors that control leukemic cell development and function strongly suggests a prominent role for epigenetic regulation in establishing and maintaining leukemic gene expression programs.
DNA Methylation
Methylation of DNA is catalyzed by DNA methyltransferases (DNMTs), which use S-adenosyl-methionine as a methyl group donor. DNA is methylated at a cytosine base that precedes a guanosine (referred to as a CpG dinucleotide) [26]. The distribution of CpG dinucleotides in the human genome is not uniform, such that there are regions of DNA enriched for CpGs, known as CpG islands. These islands are located within the promoter regions of approximately half the genes in the genome. In normal cells, the CpG island cytosines are unmethylated, in contrast to most CpG sites outside of CpG islands, which are methylated. This methylation may help maintain noncoding DNA in a transcriptionally inert state, whereas the unmethylated state of the CpG islands in gene promoters permits active gene transcription. However, the DNA methylation patterns in human cancer cells are considerably perturbed. It was established 30 years ago that cancer cells are globally DNA hypomethylated, and there is a significant decrease in the genomic methylcytosine content of malignant cells when compared to normal tissues [27, 28]. This hypomethylation of CpGs in the bulk of the cancer-cell genome has the potential to contribute to carcinogenesis by several mechanisms including: fostering chromosome instability; predisposing cells to aberrant chromosome recombination; causing re-expression of growth-promoting imprinted genes; upregulating the expression of retrotransposons (small DNA elements that move from place to place in the genome via an RNA intermediate), endogenous retroviruses and proto-oncogenes; and interfering with the normal downregulation of certain genes indirectly through hypomethylation of centromeric DNA [29].
At the same time, cancers present with gains in methylation specifically in promoter-associated CpG islands. CpG island hypermethylation and subsequent transcriptional inactivity represents an important mechanism in the pathogenesis of neoplasms. This means of tumor suppressor silencing has been shown to occur in both ALL and AML. Genes that have been frequently reported to be hypermethylated in these hematopoietic malignancies are listed in Table 14.1. Aberrant CpG island methylation in these genes leads to transcriptional shutdown and involves the recruitment of methyl-binding proteins and histonedeacetylases (HDACs) to regions near transcription start sites [30, 31].
Table 14.1
Genes silenced due to hypermethylation in acute leukemia
Gene | Protein | Type of leukemia | Protein function |
|---|---|---|---|
CALCA | Calcitonin | ALL | Peptide hormone known to participate in calcium and phosphorus metabolism |
CDH1 | E-Cadherin | ALL/AML | Calcium-dependent cell–cell adhesion protein |
CDKN1C | p57(KIP2) | ALL | Cyclin-dependent kinase inhibitor |
DAPK | Death-associated protein kinase | ALL/AML | Pro-apoptotic Serine/Threonine kinase |
ER | Estrogen Receptor | ALL/AML | DNA-binding transcription factor that regulates estrogen-induced gene transcription |
HIC1 | Hypermethylated in cancer 1 protein | AML | Transcriptional repressor |
PTPN6 | Protein tyrosine phosphatase, nonreceptor type 6 (previously known as SHP1) | ALL/AML | Tyrosine phosphatase |
WT1 | Wilms’ tumor protein | AML | Zinc finger transcription factor |
p15 INK4b | p15 | AML | Cyclin-dependent kinase inhibitor |
p73 | p73 | ALL | G1-S cell cycle control |
Posttranslational Modification of Histones
Histones are proteins that previously had the boring reputation as being the scaffold around which DNA is wrapped in order to package very long DNA strands into the nucleus. They are globular proteins with a flexible N-terminus (i.e., the tail) that protrudes from the nucleosome, which consists of a nucleosome core particle (147 base pairs of DNA wrapped around a histone octamer containing two copies each of the core histones H2A, H2B, H3, and H4) plus the region of linker DNA that stretches between nucleosomes. However, the tail ends of the histones are now realized to have a more exciting role as regulators gene expression. Histone tails are covalently modified by phosphorylation, acetylation, methylation, ubiquitination, sumoylation, and ADP-ribosylation [32]. The combination of these modifications is hypothesized to function as a cipher for chromatin–DNA interactions, forming the so-called, “Histone Code.”
Global acetylation of the histone tails serves to mark genes that are “on,” and are actively being transcribed. Enzymes with histone acetyl transferase (HAT) activity add acetyl groups to histones, while the acetyl groups are removed by specific histone deacetylases (HDACs) [33]. The acetylation of histone tails reflects a delicate balance between the activities of HATs and HDACs, which affects the openness of chromatin structure. Slight perturbations in this balance can largely influence gene expression, and restoration of this equilibrium represents a therapeutic strategy in acute leukemia. Aberrant activity of HATs and HDACs resulting in deregulated gene transcription is a feature of many cancers, including many hematologic malignancies [34]. Treatment with histone deacetylase inhibitors (HDACis) affects the expression of 2–10 % of a selective subset of genes, with the ratio of upregulated to downregulated being close to 1:1 [35]. Noticeably, normal cells are almost always considerably more resistant than tumor cells to the effects of HDACis, indicating the pivotal role epigenetic modulation plays in controlling leukemogenesis.
It was demonstrated that histones can be methylated either on their arginine or on their lysine residues [36–39]. Clues to understanding how leukemogenesis is related to disruption of normal patterns of histone methylation can be found by studying the many fusion proteins containing the lysine histone methyltransferase, MLL. A more in-depth discussion of these MLL fusion proteins can found in a separate section later within this chapter.
The histone code itself is remarkably complex and remains incompletely deciphered. Additionally, the complex interplay between histone modifications and DNA methylation results in an epigenetic pattern that precisely regulates gene transcription. In general, hyperacetylation of histones and the methylation of histone H3 on lysine 4 (K4) correlate with transcriptional activation [33]. Conversely, histone deacetylation and hypermethylation of CpG islands within gene promoter regions are associated with transcriptional inactivation (Fig. 14.4).
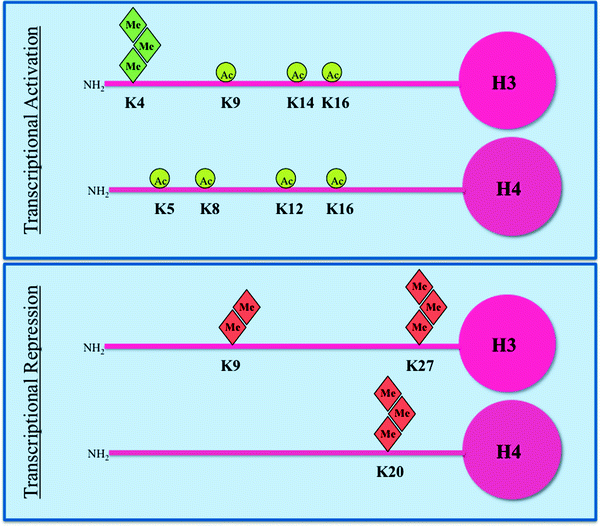
Fig. 14.4
Histone modifications in the context of transcription. Histone H3 and H4 acetylation (circles) and methylation (diamonds) are shown. In normal cells, regions that include the promoters of tumor suppressor genes are enriched in histone tail modifications associated with active transcription (top panel), such as acetylation of lysine (K) residues and trimethylation of K4 of H3. In the same cells, transcriptionally inactive regions are characterized by trimethylation of K27 and dimethylation of K9 of H3, and trimethylation of K20 of H4, which function as repressive marks (bottom panel). The situation is more complicated than depicted as there is significant crosstalk between adjacent various modifications on the same histone tail as well as between modifications on different histones
Unlike chromosomal deletions that lead to an irreversible loss of function, transcriptional repression by epigenetic mechanisms such as histone deacetylation and promoter DNA methylation can be reversed by pharmacological inhibition. Compounds which alter chromatin structure are effective at controlling leukemic cell growth. In both the laboratory and the clinic, the cytidine analogs 5-aza-2′-deoxycytidine (decitabine) and 5-azacytidine are capable of reactivating tumor suppressor genes silenced by promoter hypermethylation [40]. Incorporation of these agents into DNA leads to subsequent DNA demethylation via their irreversible inhibition of DNMTs. Both agents have proven clinical activity in myelodysplastic syndromes (MDS) [41] and are in trials in acute leukemia.
miRNAs
A new and exciting avenue of research in the molecular origins of leukemia has been launched with the discovery of microRNAs (miRNAs). MicroRNAs are endogenous ∼22 nucleotide noncoding RNAs that function as posttranscriptional silencers. They bind to complementary sequences present in coding mRNAs, usually in the 3′ untranslated regions, resulting in the degradation or interference with translation of the target mRNA (Fig. 14.5). These posttranscriptional regulators were inadvertently discovered in 1993 by Rosalind Lee, Rhonda Feinbaum, and Victor Ambros during the analysis of a Caenorhabditis elegans mutant [42]. MicroRNAs are predicted to target approximately 60 % of human protein-coding mRNAs [43] and are present in all human tissues [44]. A single miRNA can downregulate the production of hundreds of proteins [45]. miRNAs play an important regulatory role in many crucial physiological processes such as embryonic development, cellular apoptosis, and proliferation. Emerging evidence relates miRNA expression to various types of cancer, where crucial miRNAs have been shown to be either upregulated or downregulated. miRNAs function in complex regulatory networks to regulate hematopoietic differentiation and there seems to be an important correlation between the deregulation of miRNA expression and the presence of well-characterized leukemic cell abnormalities.
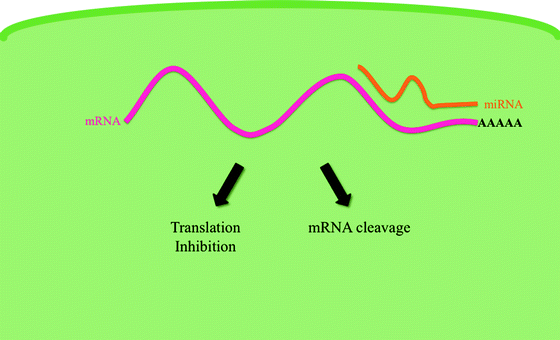
Fig. 14.5
MicroRNA mechanism of action. MicroRNAs (miRNAs) are 20–24 nucleotide RNAs that regulate eukaryotic gene expression posttranscriptionally. MiRNAs target transcripts through imperfect base pairing to multiple sites in the 3′ untranslated regions (UTRs) of the target mRNA. MiRNA–mRNA hybridization leads to translational repression or degradation of the target mRNA
Some miRNAs have been shown to function as oncogenes and their overexpression plays a part in leukemogenesis. For example, the miR-17-92 cluster encodes for seven individual miRNAs that are frequently overexpressed in MLL-rearranged AMLs [46] and ALLs [47]. The forced expression of MiR-17-92 inhibits apoptosis and increases cell viability. More importantly, the expression of this miRNA alone, and in combination with an MLL fusion, enhances the proliferation while inhibiting the differentiation of hematopoietic progenitor cells. Thus, aberrant overexpression of miR-17-92 in MLL-rearranged acute leukemias could play a role in the development of MLL-rearranged leukemias. MiR-155 has also been identified as a significant miRNA expressed by hematopoietic cells. Its expression is tightly regulated in myeloid and lymphoid cells and increases after exposure to inflammatory stimuli. MiR-155 has been implicated in hematopoietic disease in three ways: first, MiR-155 is overexpressed in patients with AML-M4 and AML-M5 (according to the French–American–British classification system of acute leukemia); second, its sustained overexpression in mouse HSCs results in a myeloproliferative disorder; and third, it has been shown to directly repress key mRNAs involved in hematopoiesis [48]. Finally, miR-196a and miR-196b are upregulated in MLL-associated AMLs and AMLs with nucleophosmin (NPM) mutations. The expression of a MLL fusion protein caused overexpression of miR-196b and knockdown of the miRNA abrogated the replating potential of bone marrow cells expressing the MLL fusion [49]. Again, forced expression of miR-196b in bone marrow cells increased proliferative capacity and survival and led to a partial block in differentiation. The common leukemogenic mechanism of action of all three oncogenic miRNAs seems to be through increasing proliferation while blocking differentiation of hematopoietic progenitor cells.
MiRNAs are also hypothesized to function as tumor suppressors and may be underexpressed in certain acute leukemias. The best characterized in the context of acute leukemia is the let-7 family, which negatively regulates the human RAS [50] and HMGA2 [51] oncogenes. Its expression is found to be decreased in core-binding factor leukemias [52] (see later section on these leukemias) and is increased in APL cells when induced to differentiate with pharmacological doses of all-trans retinoic acid [53].
There are still unresolved paradoxes concerning miRNAs and their role in promoting acute leukemia. Computational and experimental analyses suggest that each miRNA regulates hundreds of targets [45], yet genetic studies usually show that the pleiotropic phenotypes that result from the loss of a miRNA can often be suppressed by the mutation of just one particular target gene [54]. The surprisingly modest extent of miRNA-mediated target repression (which rarely exceeds twofold [45]) seems inconsequential when compared to other normal intrinsic variations in gene expression [55]. One view is that miRNAs may act as a rheostat and fine tune the expression of most of their targets. This may be particularly relevant in the development of leukemia, as even a fine-tuning of the expression of a particular protein may be sufficient to disrupt the delicate balance between proliferation and differentiation.
Protein Translation
The amount of an active protein available to carry out defined physiological functions is ultimately what governs cell growth and proliferation. Protein translation represents one important level of regulation of protein levels in cells, and protein synthesis is coupled to essential cellular events such as growth, proliferation, differentiation, and apoptosis. In normal hematopoietic cells, messenger RNA (mRNA) translation is tightly regulated at the initiation phase by signal transduction pathways that modify two proteins, ribosomal protein S6 kinase (S6K1) and eukaryotic translation initiation factor 4E (eIF4E). When activated, both of these proteins bind at the 5′untranslated region (5′ UTR) of mRNA and enhance the binding of mRNA to polysomes, leading to increased translation.
Deregulated translational control can therefore serve as a driving force for the oncogenic process. Indeed, morphological changes of the nucleolus, the site of ribosomal RNA synthesis, have been recognized as a consistent feature of cancer cells since the end of the nineteenth century [56]. mRNA translation is an extremely complex process in the eukaryotic cell and deregulation leading to oncogenesis could occur at numerous levels. Three hypotheses speculating on the mechanism by which aberrant mRNA translation could promote neoplastic transformation have been put forward [57]:
1.
The protein synthetic machinery could be stimulated to translate an excessive amount of proteins. This implies a general, nonspecific increase in global mRNA translation leading to an increase in cell size and ultimately resulting in unrestrained cell division.
2.
The translation of specific mRNAs could be enhanced or reduced. In terms of cancer, an increase in translation of “oncogenic” mRNAs or a decrease in “tumor suppressive” mRNAs could disrupt the balance of the resultant proteins in the cell.
3.
A dysfunctional ribosome could mistranslate mRNAs into proteins with gains in oncogenic function or loss of tumor suppressor capability.
In terms of leukemogenesis, current evidence supports the mechanism suggested in the second scenario. eIF4E is a proto-oncogene that enhances both mRNA export from the nucleus and mRNA translation in the cytoplasm of a subset of transcripts coding for proteins involved in cell survival and proliferation, thereby increasing their availability to the translation machinery and allowing increased protein production in the absence of increased transcription. In the cytoplasm, eIF4E recruits mRNAs to the ribosome for initiation of translation. Importantly, eIF4E differentially affects the translation efficiency of mRNAs, where mRNAs with highly structured 5′ untranslated regions (UTRs) are regulated by eIF4E, and housekeeping transcripts such as GAPDH are not. eIF4E is found in much lower amounts than other translation factors in normal cells, so that its availability, negatively regulated by 4E binding proteins (4EBPs), is a rate-limiting factor in initiating the translation of target genes. Phosphorylation of 4EBP1 following mitogenic stimuli, via the mTOR pathway, releases eIF4E, making it available for binding to eIF4G and allowing assembly of the eIF4F translation factor (eIF4E + eIF4G + eIF4A) (Fig. 14.6).
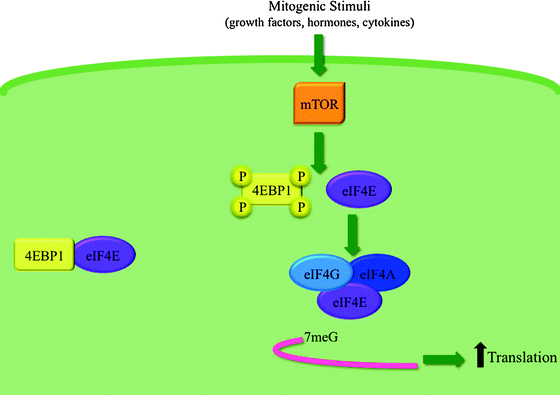
Fig. 14.6
Model of eIF4E’s role in cap-dependent translation. Under basal conditions, 4EBP1 sequesters eIF4E. Upon stimulation, mTOR phosphorylates 4EBP1 on several sites. This causes the dissociation of eIF4E from 4EBP1 freeing eIF4E to form the eIF4F translation complex, along with eIF4G and eIF4A. The eIF4F complex then recognizes and binds to the 7-methyl guanosine (7meG) cap at the 5′ end of mRNAs, resulting in an increase in cap-dependent translation
Overexpression of eIF4E is associated with oncogenic transformation in cell culture and with tumor formation, metastatic disease, and increased tumor invasion in mice. Even moderate overexpression of eIF4E promotes cellular proliferation, growth, and malignant transformation. This can be explained by the fact that the specific mRNAs targeted by eIF4E include many that are critical to cell division, cell growth, and angiogenesis, such as cyclin D1, survivin, c-myc, VEGF, Mcl-1, and bFGF-2 [58]. Overexpression of eIF4E occurs in AML and this overexpression impedes differentiation of leukemia cells. It can be speculated that eIF4E-overexpressing leukemic cells have become dependent on eIF4E for growth and survival and thereby have an addiction to eIF4E.
Several approaches to target eIF4E therapeutically have been proposed. Ribavirin is a well-characterized broad spectrum nucleoside analog with antiviral activity against a range of RNA viruses. Ribavirin targets eIF4E directly in a variety of systems, thereby inhibiting translation and/or mRNA export of transcripts that contain long and highly structured UTRs. A phase I/II clinical trial examining the efficacy of Ribavirin treatment in poor prognosis M4 and M5 AML patients demonstrated clinical activity and associated molecular responses. This provided proof of principle that targeting eIF4E leads to dramatic clinical improvements in this poor prognosis patient population [59].
In conclusion, it is becoming apparent that aberrant control of mRNA translation is an important force underlying tumorigenesis. This has important therapeutic implications, since the molecular network regulating mRNA translation is accessible for pharmacological manipulation.
Chromosomal Abnormalities
There is a long established relationship between structural genomic aberrations, especially chromosome translocations and inversions, and acute leukemia. Specific cytogenetic abnormalities are often uniquely associated with clinically distinct subsets of the disease (Table 14.2). Identifying the DNA sequences surrounding the chromosomal breakpoints has had an enormous impact on our understanding of leukemogenesis, carcinogenesis in general, and the normal cellular functions of the protein products of the involved genes.
Table 14.2
Cytogenetic abnormalities in acute leukemia
Cytogenetic abnormality | Resultant fusion protein | Malignancy (subtype) | Mechanism(s) of action |
|---|---|---|---|
t(15;17) (q22;q21) (and other chromosomal translocation involving chr. 17) | PML–RARA (and other X–RARA fusions) | AML (M3) | Inhibition of normal RARA function as an inducer of gene expression |
t(8;21)(q22;q22) | RUNX1–CBFA2T1 (previously known as AML1–ETO) | AML (M2) | Inactivation of normal RUNX1–CBFB transcription factor |
inv16(p13q22) t(16;16)(p13;q22) | CBFB–MYH11 | AML (M4eo) | Inactivation of normal RUNX1–CBFB transcription factor |
t(11q23) | MLL-various | ALL/AML (M4 and M5) | Aberrant activity of MLL transcription factor resulting in sustained expression of HOX genes |
t(19;22)(q34;q11) | BCR–ABL | ALL (B-precursor)/AML | Increased proliferation, inhibition of apoptosis and cell adhesion abnormalities |
t(12;21)(q13;q22) | ETV6–RUNX1 | ALL (Pre-B) | Inactivation of normal RUNX1–CBFB transcription factor |
t(1;19)(q23;p13) | E2A–PBX1 | ALL (Pre-B) | Not well defined |
A chromosome translocation is a structural abnormality resulting from the exchange of pieces between two nonhomologous chromosomes. Reciprocal translocations are those in which there is no obvious overall loss of chromosomal material, and they probably result from a failure to properly repair DNA double strand breaks (Fig. 14.7). A number of recurrent reciprocal translocations are frequently found in acute leukemia. Specific translocations are often consistently associated with specific subtypes of leukemia, providing strong support that they represent causal events during leukemogenesis in developing hematopoietic cells. For example, the t(15;17) is found only in patients with APL and not other forms of leukemia, while the t(1;19) is found only in the leukemic cells of patients with B-cell precursor acute lymphoblastic leukemia (ALL). However, there are exceptions to this phenomenon. For instance, the t(4;11) translocation occurs in patients with both AML and ALL. Chromosomal inversions are also seen in acute leukemia. An inversion occurs when a single chromosome undergoes breakage and rearrangement within itself.
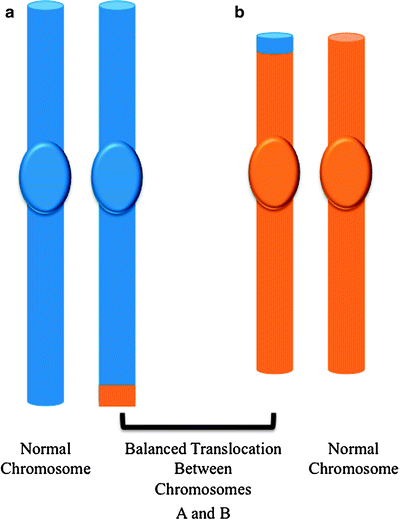
Fig. 14.7
Reciprocal balanced chromosmal translocations. A translocation arises when an exchange of chromosomal material takes place between two different chromosomes. If the translocation is balanced, the pieces of the chromosomes are rearranged but there is no loss or gain of genetic material
Identifying the genes disrupted by breakpoints in translocations and inversions provided the first mechanisitic insights into understanding why chromosome abnormalities can be leukemogenic.
X–RARA Rearrangements
Acute promyelocytic leukemia (APL) is a subtype of acute myelogenous leukemia, representing 5–8 % of AML cases in adults. At the genetic level, APL is characterized by a specific chromosomal rearrangement between the retinoic acid receptor alpha (RARA) on chromosome 17, and a number of partners. The majority of patients (98 %) presents with the 15;17 translocation, t(15;17), which results in a fusion of RARA with the promyelocytic leukemia (PML) gene on chromosome 15 [60, 61]. In normal cells, the retinoic acid receptors (RARs) heterodimerize with the retinoid X receptors (RXRs) to form a transcriptional complex. In the absence of ligand, all-trans retinoic acid (RA), a vitamin A derivative, the heterodimers are found bound, along with inhibitory corepressor molecules, to the retinoic acid response elements (RAREs) of target genes. Treatment with ligand causes a release of the corepressors and a concurrent recruitment of coactivators, leading to the initiation of transcription of genes important for stimulating myeloid differentiation and regulating the cell cycle.
In APL cells, the prevailing view is that the leukemic effects of the resulting chimeric protein, X–RARA, are due to its function as a dominant negative inhibitor of normal retinoid receptor function. The chimera locates to promoters normally regulated by RARA, aberrantly recruits corepressor proteins, and thereby inhibits the RARA-mediated gene expression. Therapeutic doses of RA can circumvent the differentiation block [62].
To date, seven different chromosomal translocation partners have been identified in patients with APL (Fig. 14.8), and, as mentioned previously, all involve the RARA gene on chromosome 17q21. The N-terminal fractions contributed by the fusion partners donate additional dimerization domains. These acquired dimerization domains, together with the DNA-binding domain of RARA, are required for the oncogenic effect of the fusion proteins and promote formation of chimeric receptor homodimers and provide additional corepressor binding domains. The resulting X–RARA fusion proteins have been shown to recruit the HDAC, NCOR1, and NCOR 2 (NCoR) complex, DNMT1, DNMT3A, repressive histone methyltransferases, and polycomb group proteins.
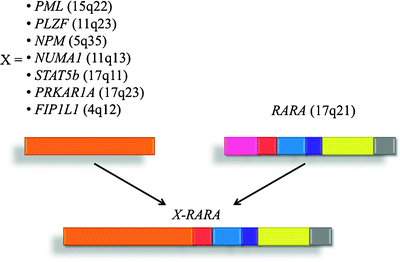
Fig. 14.8
Identified chromosomal translocations associated with APL. Acute promyelocytic leukemia (APL) is the result of chromosomal rearrangements in which part of the retinoic acid receptor alpha (RARA) gene is linked to the amino-terminal domains of seven genes (X) that encode different nuclear proteins. All of the translocations produce a chimeric protein (X–RARA) that disrupts the normal function of RARA and arrests the maturation of myeloid cells at the promyelocyte stage. The most common translocation, t(15;17), involves a break in chromosome 15 and disrupts the promyelocytic leukemia (PML) gene, producing the PML-RARA fusion
However, disruption of the normal RARA signaling pathway does not solely account for the pathogenesis of APL. As mentioned previously, the differentiation block mediated by the X–RARAs is necessary, but not sufficient to cause APL. This suggests that a second transformative event is required for full neoplastic development. Mutations in oncogenes such as FLT3 can cooperate with the fusions to provide the second hit necessary to generate leukemia. The presence of these second mutations might also explain why APL cannot be cured by differentiation therapy with ATRA alone, but is highly curable by combinations of ATRA and cytotoxic chemotherapy. Additionally, some translocations generate the reciprocal RARA–X fusion genes which can result in the coexpression of their transcripts in leukemic blasts [63–65]. Important roles in oncogenesis are now being identified for the coexpressed RARA–X reciprocal fusion proteins [63, 64, 66, 67]. Additionally, the partner proteins in APL have important growth-regulatory roles in normal myeloid cells. It may be that the loss of one allele of the partner protein combined with the effects of the X–RARA or reciprocal RARA–X fusion protein, which compromises normal partner protein function, contribute to the development of leukemia.
Core-Binding Factor Rearrangements
Core-binding factor (CBF) leukemias refer to a subset of AML bearing t(8;21)(q22;q22) and inv(16)(p13q22). These genomic rearrangements are characterized by disruption of the RUNX1 gene at 21q22 and the CBFbeta (CBFB) gene at 16q22, respectively. These are two of the most prevalent cytogenetic subtypes of AML. Native RUNX1 and CBFB form a heterodimeric transcription factor [68] required for the development of definitive hematopoiesis, including macrophage colony-stimulating factor (CSF) receptor [69], granulocyte–macrophage CSF [70], myeloperoxidase [71], and interleukin-3 [72]. However, the normal function of the heterodimer is abrogated in the CBF leukemias.
RUNX1–CBFA2T1
The chromosomal translocation t(8;21) fuses the DNA-binding runt domain from RUNX1 (previously known as AML1) to almost the entirety of the CBFA2T1 (previously known as ETO) repressor protein [73]. An early hypotheses for the pathogenic role of the resulting fusion protein, RUNX1–CBFA2T1 postulating that it functions as a transcriptional repressor, was derived from models based on the archetypical leukemia-associated transcription factor, PML–RARA discussed earlier and are supported by several lines of evidence:
1.
2.
However, recent evidence has suggested that the model of RUNX1–CBFA2T1’s leukemogenic effect through repressive transcriptional activity needs to be broadened. Microarray data shows that RUNX1–CBFA2T1 activates as many genes as it represses, and that it regulates genes not normally under the control of wild-type RUNX1. Many of the genes upregulated by RUNX1–CBFA2T1, for example JAG1 [81], play a role in promoting stem cell renewal. Furthermore, RUNX1–CBFA2T1 is a potent transcriptional activator of the anti-apoptotic BCL2 gene [82]. Thus, RUNX1–CBFA2T1 expression may result in leukemia by two opposing effects on transcription; by simultaneously repressing tumor suppressor genes while upregulating genes that drive expansion or prolong the survival t(8;21) early multipotent progenitors.
Finally, an alternative explanation for the pathogenesis of t(8,21) acute myeloid leukemia is the loss of function of lineage-program-transcription factors [83]. These transcription factors strictly control gene expression during hematopoiesis, and instruct a precursor cell to commit to a certain differentiation program by initiating expression of a characteristic set of lineage-specific target genes in response to diverse signals [84]. There are four central hematopoietic transcription factors that are functionally repressed by RUNX1–CBFA2T1 through protein–protein interactions: PU.1 [85], C/EBPA [86], GATA-1 [87], and the E proteins [88]. These inhibitory interactions with RUNX1-CBF2AT1 may expand stem cell pools by promoting stem cell renewal and blocking commitment to the various lineages. The expanded stem cell pool may then be primed for leukemic transformation through acquisition of additional mutations. The development of these secondary mutations is again facilitated by RUNX1–CBFA2T1 expression through the fusion protein’s ability to repress of DNA repair genes [81].
CBFB–MYH11
Inversion of chromosome 16, inv(16), or the related t(16;16) results in the fusion of the core-binding factor beta gene CBFB with MYH11, the gene encoding smooth-muscle myosin heavy chain. The resulting fusion gene, CBFB–MYH11, encodes the chimeric protein CBFB–MYH11, which is composed of the heterodimerization domain of CBFB fused to the C-terminal coiled-coil domain from MYH11. Studies suggest that CBFB–MYH11 has a higher affinity for RUNX1 than wild-type CBFB [89], and thus the fusion dominantly suppresses the function of the transcription factor composed of RUNX1 and CBFB, leading to the impairment of hematopoiesis. It has been shown in mice that CBFB–MYH11 is necessary but not sufficient for leukemogenesis [4], with additional cooperating mutations being required, possibly to confer a proliferative and/or survival advantage [90].
Furthermore, there are data suggesting that CBFB–MYH11 has activities independent of RUNX1/CBFB repression. CBFB–MYH11 expression has been shown to reduce levels of C/EBPA protein, which is crucial for normal granulopoiesis, without affecting mRNA levels, but rather by the translational inhibition of C/EBPA through the induction of calreticulin, a ubiquitous protein with calcium storage and chaperone function [91]. Additionally, microarray analyses have determined that CBFB–MYH11 expression results in a significant upregulation of many genes, including those involved in DNA replication, cell cycle regulation, and proliferation [92], again indicating that CBFB–MYH11 has RUNX1/CBFB repression independent activities that may contribute to leukemogenesis.
MLL Fusions
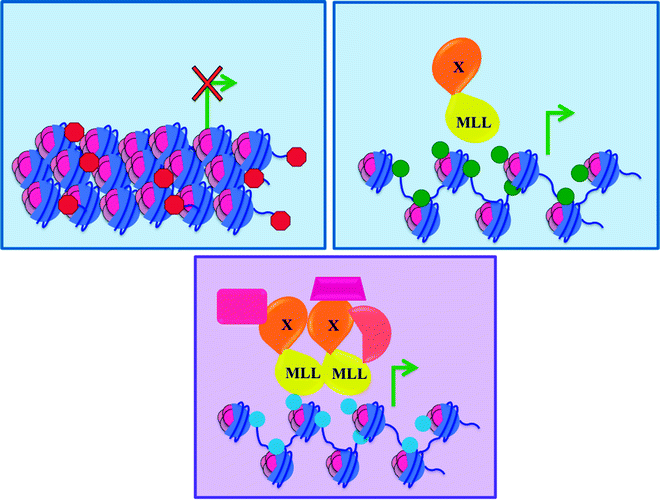
The MLL (mixed lineage leukemia) gene is located on chromosome 11q23, and fusions involving this gene are seen in both de novo and therapy-related acute myeloid and lymphoid leukemia. MLL is a large DNA-binding protein ubiquitously expressed in hematopoietic cells, including stem and progenitor populations. Using chromatin immunoprecipitation (ChIP) analysis, MLL has been found to be associated with a subset of transcriptionally active human promoters [93, 94] and with RNA polymerase II, suggesting that MLL has a specific role in the regulation of transcription. A SET domain is located at the carboxy-terminal of MLL, and this domain mediates methylation of histone H3 lysine 4 (H3K4), a histone modification which is associated with transcription at active gene loci [95]. Importantly, in mammals, MLL positively regulates the expression of the homeobox (HOX) genes. HOX genes are transcription factors that participate in the development of multiple tissues, including the hematopoietic system.
Some of the roughly 50 characterized MLL fusion partners (FPs) can be grouped into families based on cellular localization and function (Table 14.3) [96, 97]. All identified MLL fusions contain the first 8–13 exons of MLL and a variable number of exons from the FP gene. Furthermore, another type of MLL rearrangement, MLL–PTD (partial tandem duplication), is a result of internal tandem duplication of select exons. MLL mutant proteins are always in-frame chimeras that reside in the nucleus, regardless of whether the fusion partner is normally nuclear or cytoplasmic in origin. Despite the large number and functional diversity of the fusion partners, there are some common principles that can be applied to all MLL fusions [96]. First, all MLL fusions retain its amino-terminal domains required for the association of MLL with chromatin, so the fusion proteins are still able to bind DNA. Second, expression of the amino-terminal region of MLL (the region retained in MLL fusions) alone does not lead to myeloid transformation [98], indicating that the fusion partners make critical contributions to the oncogenicity of MLL fusions [99]. Third, MLL fusion proteins enforce continuous expression of the HOX genes, HOXA9 and MEIS1, which are normally downregulated during hematopoietic differentiation. This sustained induction appears to be critical for leukemogenesis, since forced expression of HOXA9 and MEIS1, in the absence of an MLL fusion, immortalizes hematopoietic progenitors in vitro and results in AML development in transplanted mice [100, 101].
Table 14.3
Classification of MLL fusion partners
Group | MLL fusion partner | Location | Function |
|---|---|---|---|
1 | AF4, AF9, AF10, ENL, ELL | Nuclear | Putative DNA-binding proteins |
2 | CBP, p300 | Nuclear | Histone acetyltransferases |
3 | AF1P, AF6, AFX, EEN, EPS15, GAS7, LARG | Cytoplasm | Presence of coiled-coil oligomerization domain |
4 | SEPT2, SEPT5, SEPT6, SEPT9, SEPT11 | Cytoplasm | Septin family, interact with cytoskeletal filaments, have a role in mitosis |
5 | N/A | N/A | MLL partial tandem duplication of exons 5–11 (MLL/PTD) |
MLL fusions are hypothesized to disrupt normal gene expression patterns, especially those of the HOX genes, maintained by wild-type MLL. However, discerning one unifying mechanism of leukemogenesis by MLL fusion proteins is difficult due to the heterogeneity of the MLL fusion partners. The putative mechanisms of MLL fusion-induced transformation may be more easily characterized by dividing the fusion partners into two broad classes based on their normal cellular localization (either nuclear or cytoplasmic). The fusion partners that are nuclear have been associated with various aspects of transcriptional regulation. Therefore, the fusions involving these proteins may lead to transcriptional deregulation of the HOX genes through alterations in the histone modification pattern and chromatin structure (Fig. 14.9). An early insight into this possibility was provided by the characterization of the nuclear MLL–CREBBP fusion [102]. CREBBP (also known as CBP) is a well-characterized global transcriptional activator with intrinsic histone acetyltransferase (HAT) activity. Structure–function analysis demonstrated that inclusion of the portion of CREBBP that contains its HAT domain in the fusion is required for full in vitro transformation and is sufficient to induce the leukemic phenotype in vivo [103]. These data suggest that the leukemic effect of MLL–CREBBP results from the combination of the chromatin association and modifying activities of CREBBP with the DNA-binding activities of MLL. Additionally, although the SET domain, which mediates MLL’s H3K4 methyltransferase activity, is consistently lost in all of the MLL fusions, several MLL fusion partners, for example, AF4, AF9, AF10 and ENL [104], associate with the DOT1L histone methyltransferase that methylates lysine 79 residues in histone H3 (H3K79). Methylation of H3K79 is also associated with positive transcriptional regulation [105]. As different methylation marks may positively control transcription in unique ways [106], the replacement of H3K4 activity in wild-type MLL with H3K79 activity in the MLL fusion complex could perturb transcriptional control. In the case of the MLL–PTD, where the SET domain is maintained, the characterization of a mouse model of Mll–PTD demonstrated that MLL–PTD facilitates histone H3K4 trimethylation as well as H3/H4 acetylation within target Hox gene promoters [107]. This provides further evidence for an epigenetic mechanism as the underlying cause of Hox overexpression.