Fig. 20.1
Hematopoiesis. The current model of hematopoiesis is depicted below. In this model hematopoietic cells are derived from a small number of HSCs that progress through a series of progenitors that are characterized by progressive restriction of the ability to proliferate and generate different hematopoietic progenitors. HSC hematopoietic stem cells, CLP common lymphoid progenitor, CMP common myeloid progenitors, G granulocytes, M monocytes, P-B pre-B cells, GMP granulocyte macrophage progenitor, myelo-erythroid progenitor
Hematopoiesis is governed by a complex set of intrinsic coordinated gene expression processes that can be strongly influenced by a variety of environmental factors, which include cytokines, chemokines, direct cell contact with other cell types, and tissue oxygen levels. Particularly important is the bone marrow microenvironment, which is very complex with a wide array of growth, differentiation, migration, and other signals constantly bathing the HSCs and progenitors and influencing their growth and development [4].
20.2 Myelodysplasia and Acute Leukemia
Given the tremendous amount of cell turnover and the complex intracellular and environmental regulation of hematopoiesis, it is not surprising that there are a number of hematopoietic disorders ranging from low blood counts (cytopenias) to myelodysplasia (MDS) and acute leukemia. MDS is a spectrum of disorders of blood production, typically characterized by anemia, neutropenia, or thrombocytopenia and a tendency to transform into acute leukemia [5]. The natural history of MDS ranges from indolent and stable for years to rapidly progressive with death occurring within months from infections, bleeding or evolution to acute leukemia. Approximately 15,000 people, most in their 60s and older, develop MDS in the USA each year. The known environmental associations with MDS include exposure to benzene, radiation, and prior chemotherapy treatment of other malignancies but in most patients, there is not an obvious cause. The treatment of MDS depends on whether it is considered low risk or high risk for progression and death. Low risk patients may require no treatment or may need transfusions, growth factor administration, or treatment with hypomethylating agents such as azacytidine, which broadly effect gene expression patterns. High risk MDS is treated with allogeneic stem cell transplantation if the patient is fit enough to be able to tolerate this aggressive form of therapy.
Acute leukemia comes in two major versions, acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). These are likely derived from malignant transformation of myeloid and lymphoid hematopoietic progenitors, respectively, although some evidence indicates that in some cases, leukemic transformation may occur at the level of the HSC. Approximately 14,000 people develop AML each year and it typically effects people in their 60s or older although it can develop at any age. AML is also a wide range of diseases, driven by a variety of different molecular processes [6]. Most patients with AML are treated with chemotherapy and patients deemed at high risk for relapse but otherwise fit are further treated with an allogeneic stem cell transplant in an effort to reduce the chances of relapse. Overall about 30–40 % of patients with AML can be cured; however there is great variation depending on the genetic abnormalities in the AML and the age and fitness of the patient. In patients with high risk cytogenetic features, <5–10 % of patients can be cured [7]. In contrast, ALL occurs more commonly in children although it can occur in adults of any age as well [8]. Approximately 6,000 people are diagnosed per year with ALL, and in childhood the majority of patients are now cured with prolonged courses of multiple chemotherapy agents. However, in adults the cure rate is much lower, largely due to relapse of the disease. Patients deemed at high risk for relapse after treatment with chemotherapy alone are treated with allogeneic stem cell transplantation.
20.3 The Effects of Alcohol on Hematopoiesis
Heavy and prolonged alcohol use has long been known to cause cytopenias including anemia, leukopenia, and thrombocytopenia. In a group of heavy alcohol users, anemia occurred in around half and thrombocytopenia and leukopenia were also common [9]. A variety of abnormalities in red cell size and structure have been observed including increased RBC cell volume and the development of abnormal RBCs including macrocytes, stomatocytes, and others. Morphologic analysis of bone marrow samples revealed vacuolization and other abnormalities indicative of toxic effects [9–11]. In addition to suppression of red cells, platelet, and neutrophils, alcohol may also suppress B-cell development and thereby impact immunity [12]. Ethanol and its primary reactive metabolite acetaldehyde (Fig. 20.2) also significantly suppress neutrophil and macrophage function and increase intracellular reactive oxygen species (ROS) [13].
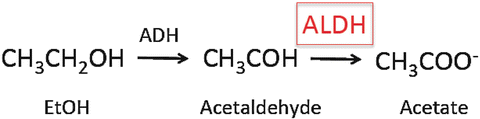
Fig. 20.2
Role of ALDHs in ethanol metabolism. ALDH aldehyde dehydrogenase, ADH alcohol dehydrogenase
The toxic effects of alcohol on hematopoiesis are compounded by other factors including concomitant medications and illnesses. For example, in a murine model of HIV, chronic administration of ethanol for 7 weeks resulted in a substantial reduction in erythroid and myeloid progenitors, which was worsened by expression of HIV proteins in hematopoietic cells or by treatment with the HIV drug azidothymidine (AZT). This study indicates that ethanol ingestion in HIV-1-infected individuals, particularly those on antiretroviral drugs, might increase bone marrow toxicity and contribute to HIV-1-associated hematopoietic impairment [14]. In part this may be due to the adverse effects of AZT on mitochondrial DNA replication that may be additive to the known deleterious effects on other aspects of mitochondrial function [15, 16]. Similarly, in mice exposed to ethanol and benzene, another marrow toxin, a variety of abnormalities in RBC production were noted [17].
The cytopenias from alcohol have been investigated in both animal models and in human in-vitro models of hematopoietic cell growth. Laboratory measurements of hematopoietic progenitor cell growth such as colony forming (CFU) assays demonstrate sensitivity to ethanol and an even more marked inhibition by acetaldehyde. Suppression of erythroid progenitors occurred more readily than suppression of myeloid progenitors [18], which may partially explain why anemia is more common than leukopenia in heavy alcohol users. In contrast, suppression of primitive multipotent progenitors was relatively minimal with concentrations of ethanol and acetaldehyde that suppressed more committed myeloid and erythroid progenitor cells. These findings indicated that suppression of hematopoiesis by alcohol occurred primarily at the level of committed hematopoietic progenitors, there was differential sensitivity of myeloid and erythroid progenitors to alcohol and that both ethanol and acetaldehyde played roles in the development of cytopenias following alcohol use [18]. The effects of ethanol specifically on platelet formation have also been investigated. Immature megakaryocyte progenitors as measured in CFU assays had a dose dependent suppression by ethanol. In contrast, acetaldehyde did not inhibit more mature megakaryocytes unless very high concentrations were used while they retained sensitivity to ethanol [19, 20].
In addition to suppressing baseline hematopoiesis, chronic ethanol may also block the dynamic response of hematopoiesis to demands such as infections. In a murine model, long-term ethanol exposure blocked the increase in blood granulocyte counts normally associated with intrapulmonary inoculation with S. pneumoniae. This suppression was associated with a significant decrease in bone marrow myeloid progenitor cell proliferation and was accompanied by increased STAT3 phosphorylation. In addition, ethanol increased G-CSF induced activation of the STAT3–p27Kip1 pathway in a mouse hematopoietic progenitor cell line while inhibiting growth proliferation and inducing cell cycle arrest [21]. Similarly, in a murine model of pneumonia, ethanol exposure impaired the pneumococcal-induced increase in neutrophil recruitment into the alveolar space, decreased bacterial clearance from the lung, and increased mortality from pneumonia. At least part of this was due to impaired proliferation of early marrow hematopoietic progenitors [22]. In addition to these direct effects of alcohol on hematopoiesis, indirect effects may occur as well. For example, ethanol has been shown to decrease the production of growth factors from T-cells which are important for normal hematopoiesis [23]. The exact molecular mechanisms by which ethanol and acetaldehyde suppress hematopoiesis remain unknown. Several lines of evidence implicate the formation of reactive aldehyde adducts with proteins, DNA, and other cellular constituents as part of the mechanism. In immunohistochemical analysis of erythroid cells from blood and marrow from patients with heavy ethanol use, acetaldehyde adducts occurred on both the cell membrane and intracellularly [24, 25].
Together, these findings demonstrate that ethanol and its metabolite acetaldehyde have wide ranging direct and indirect effects on hematopoiesis, immunity, and cell function that in part be mediated by adduct formation by acetaldehyde.
20.3.1 Alcohol and the Risk of MDS and Leukemia
Given the wide-spread deleterious effects of ethanol on the hematopoietic system, associations with the development of MDS and leukemia have been investigated in a number of epidemiologic and laboratory studies. In a case controlled Japanese study, alcohol consumption was associated with approximately a twofold increase in the risk of MDS and this risk was dose dependent [26]. An Italian population based case control study demonstrated that there may be an increased incidence of AML in people consuming high levels of alcohol [27]. A hospital case control study in Shanghai also demonstrated that alcohol may be a possible risk factor for the development of AML [28]. In contrast, in a US population based case control interview study, there was a mildly increased risk for MDS and leukemia in alcohol users, which did not reach statistical significance [29]. In patients in the USA with AML and known pre-existing MDS, no association was found with alcohol use [30]. Similarly, in a large cohort study from the Netherlands no association with the development of AML was observed with ethanol while the association with MDS was not examined [31]. Together, these epidemiologic studies are inconsistent so that it currently remains unclear whether there is an association between alcohol use and development of MDS and leukemia.
In laboratory studies, there is more consistent and clear evidence that ethanol and acetaldehyde can lead to AML development. In mice genetically engineered to be deficient in the acetaldehyde metabolizing enzyme Aldh2 and the DNA repair enzyme Fancd2 (which is responsible in part for repairing acetaldehyde induced DNA crosslinks), ethanol exposure led to bone marrow failure and spontaneous AML development [32]. In follow-up studies, Aldh2 and Fancd2 deficient mouse HSCs were found to be the more sensitive to damage by reactive aldehydes than more committed progenitors. This further confirmed that reactive aldehydes from both exogenous and endogenous sources might play an important role in HSC biology. In addition, these studies indicated that congenital bone marrow disorders with a propensity to bone marrow failure and AML development, such as Fanconi’s anemia (which is caused by deficiency of Fancd2), might also be caused by excess accumulation of reactive aldehydes [33]. In support of this possible explanation for human diseases of the marrow, Japanese Fanconi’s anemia patients with polymorphisms in ALDH2 that affected enzymatic function had accelerated progression to bone marrow failure compared to their counterparts with normal aldehyde dehydrogenase (ALDH) activity [34]. These studies show that the affect of alcohol on hematopoietic cells including HSCs may be complex and at least in part be dependent on the presence or absence of deficiencies in DNA repair enzymes like FancD2 and acetaldehyde metabolizing enzymes including the ALDHs.
20.4 ALDH and HSCs
As discussed elsewhere in this volume, ALDHs play a key role in metabolizing alcohol (Fig. 20.2). ALDH activity is higher in HSCs compared to more mature progenitors and most differentiated hematopoietic cells. Based on this distinction, a fluorescent ALDH substrate termed Aldefluor has been developed that is useful in the isolation of human hematopoietic cells as well as stem cells in other tissues and species [35]. In studies investigating the biology of ALDHs in HSCs and other hematopoietic cells, Aldefluor was found to be primarily a substrate for the ALDH1A1 isoform and to a lesser extent the ALDH3A1 isoform while other studies have also found it is a substrate for ALDH2 as well [36, 37]. Surprisingly, however, loss of ALDH1A1 in hematopoietic cells did not lead to any clear phenotype [38]. However, loss of ALDH1A1 led to an increase in expression in the ALDH3A1 isoform, presumably as a compensatory mechanism [36]. When mice deficient in both ALDH1A1 and ALDH3A1 were examined, there were a wide range of hematopoietic defects under baseline conditions including a block in B-cell development as well as abnormalities in cell cycling, intracellular signaling, and gene expression along with reduced numbers of HSCs. Intriguingly, hematopoietic cells from these mice had increased ROS and reduced metabolism of the reactive aldehyde 4-hydroxynonenal and sensitivity to DNA damage from these agents [36]. Together these findings further support a role for ALDHs in HSCs in metabolizing endogenous and exogenous reactive aldehydes such as acetaldehyde derived from ethanol.
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
Fetal Alcohol Exposure Increases Susceptibility to Carcinogenesis and Promotes Tumor Progression in Prostate Gland
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


