Fig. 21.1
Tumor incidence in two models of HCC and EtOH consumption. (a) Twenty-one-day-old male C57BL/6 or B6C3 mice were injected with vehicle (Veh; 100 μL sterile olive oil) or DEN (5 mg/kg, C57BL/6; 3 mg/kg B6C3). At 35 weeks mice were weaned onto either 10 %/20 % (v/v) alternate day ethanol in drinking water (EtOH) or drinking water alone (H2O) until 42 weeks. Tumors formed in only 60 % of C57BL/6 mice, an effect that was mildly suppressed by EtOH (44.4 %). Tumor incidence was 85.7 % and 92.9 % respectively for B6C3 mice injected with DEN or DEN plus EtOH regimen, which was significantly higher than C57BL/6 mice. *p < 0.05 vs. C57BL/6 EtOH DEN (b) Tumors were much larger in B6C3 DEN mice than C57BL/6 mice, an effect exacerbated by EtOH in B6C3 animals alone. No tumors were identified in Veh-injected mice
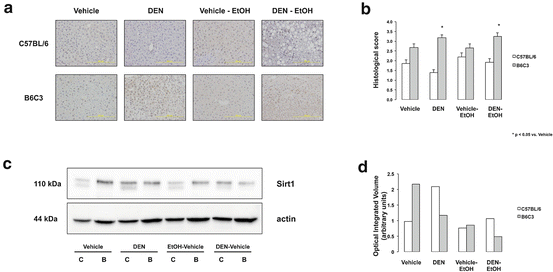
Fig. 21.2
Sirt1 expression in mouse models of HCC and EtOH consumption. (a) Representative Sirt1 IHC of hepatic sections (×200 magnification) from C57BL/6 and B6C3 mice injected with or without DEN and placed on EtOH or drinking water alone. Sirt1 expression was higher in B6C3 mice vs. C57BL/6 mice. DEN administration caused a significant increase in Sirt1 expression in B6C3 mice only. No effect due to EtOH was observed. (b) Data from Sirt1 IHC scoring in graph format. *p < 0.05 vs. Veh-injection (B6C3 mice). (c) Representative Western blot for Sirt1 expression from pooled liver lysates. (d) Quantitative of bands following normalization with β-actin showed increased Sirt1 expression in B6C3 Veh-injected mice, in contrast to IHC findings. Likewise, increased Sirt1 expression was detected in DEN-injected C57BL/6 mice
EtOH treatment on Sirt1 expression in HCC cell lines: To validate expression of EtOH-metabolizing enzymes, Western blot analysis was performed on lysates from HepG2, VA-13, E47 and VL-17a cell lines. As expected, ADH expression was strongest in VA-13 and VL-17a cells, and CYP 2E1 expression was strongest in E47 and VL-17a cells, although weak expression was detected in HepG2 and VA-13 cell lines (Fig. 21.3a).
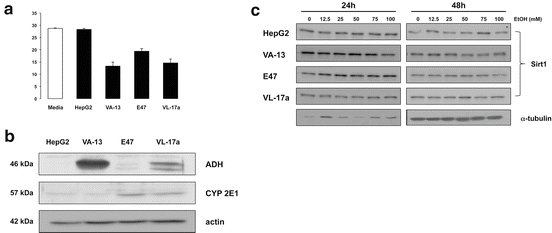
Fig. 21.3
EtOH metabolism and effect on Sirt1 expression in HCC cell lines varying in EtOH metabolism capacity. (a) Representative Western blots from HepG2, VA-13, E47 and VL-17a HCC cell lines. ADH expression was highest in VA-13 and VL-17a cells, with minimal expression in HepG2 and E47 cell lines. ALDH was expressed highest in E47 and VL-17a cell lines and CYP 2E1 had highest expression observed in E47 and VL-17a cells. (b) Sirt1 expression from HCC cell lines treated with 0–100 mM of EtOH for 24 and 48 h. Western blots for Sirt1 showed no effect on Sirt1 expression was seen in HepG2 cells; however, decreases were seen in VA-13 (24 and 48 h), E47 (48 h), and VL-17a cell lines (48 h)
Cells were treated with 50 mM EtOH for 24 h to evaluate ethanol-metabolizing capacity. HepG2 cells had minimal EtOH-metabolizing capacity while ADH-expressing VA-13 and VL-17a cells had highest EtOH-metabolizing capacity, and E47 cells displayed intermediate capacity (Fig. 21.3b).
Treatment of cells with increasing concentrations of EtOH (0–100 mM, 24 or 48 h) demonstrated no significant change in Sirt1 protein expression in HepG2 cells (Fig. 21.3b). However, treatment of VA-13 (24 and 48 h), E47 (48 h), and VL-17a cell lines (48 h) demonstrated decreased Sirt1 expression in a dose-dependent manner, increased effect (decreased Sirt1 protein) being detected at increasing doses of EtOH (Fig. 21.3c).
Effects of EtOH and Sirt1 activity on HCC cell line proliferation: To determine if changes in Sirt1 expression affect proliferation of HCC cells lines with varying capacities to metabolize EtOH, HepG2, VA-13, E47, and VL-17a HCC cells were treated with or without EtOH (25 mM) in the absence or presence of a Sirt1 activator (CAY10591, 5 μM) or inhibitor (EX-527, 5 μM) (24 or 48 h). HepG2 cells, which have low EtOH-metabolizing capacity, had no significant change in proliferation at 24 h with the exception of cells treated EX-527, which significantly increased proliferation. At 48 h, there was a significant reduction in cell proliferation following CAY10591 treatment, while cells pretreated with EX-527 (Fig. 21.4a) exhibited significant increases in proliferation. We next treated VA-13 with the same regiment and found again, reduction in proliferation with Sirt1 agonist at both 24 and 48 h; however, there were no significant changes in other treatment groups, including the Sirt1 inhibitor group (Fig. 21.4b). Treatment of the CYP 2E1-only overexpressing cell line, E47, led to increased proliferation at 48 h with EX-527 treatment; however, no significant decrease in proliferation was measured with CAY10591 treatment at either time point (Fig. 21.4c). We then examined CYP 2E1 and ADH-expressing VL-17a cells and measured significant differences in proliferation between EX-527-treated cells and controls at 48 h, as well as between EX-527- and CAY10591-treated cells. Additionally, the CYP 2E1 and ADH-expression VL-17a cells were the only cells that had increased proliferation in the presence of EtOH treatment (Fig. 21.4d).
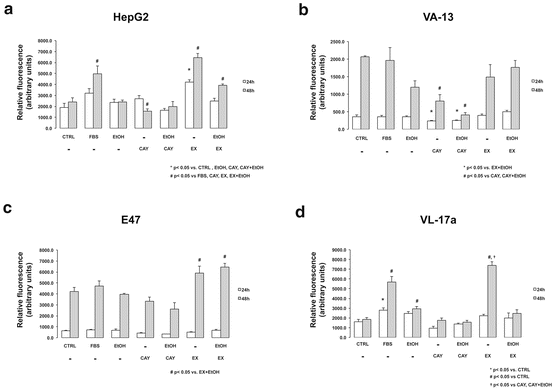
Fig. 21.4
Effect of Sirt1 modulation on proliferation in HCC cell lines with varying EtOH metabolism capacity. Cell lines were treated with or without 1 % FBS (FBS), 25 mM EtOH (EtOH), 5 μM CAY 10591 (CAY), 5 μM EX-527 (EX) or no treatment (CTRL) (a) HepG2 had no significant changes at 24 h except for treatment with the Sirt1 inhibitor EX-527, which significantly increased proliferation. At 48 h, there was a significant reduction in proliferation with CAY treatment, and significant increases in proliferation with EX. *p < 0.05 vs. CTRL, EtOH, CAY, CAY + EtOH # p < 0.05 vs. FBS, CAY, EX, EX + EtOH (b) We next treated with VA-13 and observed reduction in proliferation with CAY at both 24 and 48 h; however, there were no significant changes in other treatment groups, including EX. *p < 0.05 vs. EX + EtOH # p < 0.05 vs. CAY, CAY + EtOH (c) Treatment of the CYP 2E1-only overexpressing cell line, E47, showed increased proliferation at 48 h with X treatment; however, no significant decrease in proliferation was seen with CAY at either time point. # p < 0.05 vs. EX + EtOH (d) We then examined VL-17a cells and detected significant differences in proliferation between EX-treated cells and CTRL cells at 48 h, as well as between EX- and CAY-treated cells. Additionally, VL-17a cells were the only cells that had increased proliferation in the presence of EtOH treatment *p < 0.05 vs. CTRL # p < 0.05 vs. CTRL † p < 0.05 vs. CAY, CAY + EtOH
21.4 Discussion
We report increased Sirt1 expression in nuclei of hepatocytes in animals with significant liver tumor burden (DEN-injected) compared to Veh-injected animals on a B6C3 background (Fig. 21.1a, b); however, EtOH administration did not further enhance Sirt1 staining. These findings are somewhat paradoxical, as Sirt1 expression has been correlated to increased liver tumor progression/poor prognosis in humans; however, Sirt1 expression and activity are impaired by EtOH metabolism [18, 21]. Thus, future studies to assess acetylation of downstream Sirt1 targets such as NF-κB and SREBP-1c in HCC, including HCC in the setting of EtOH abuse would be of interest.
HCC is an increasingly diagnosed disease state in the liver for which alcohol is a leading risk factor in developed countries [33]. The deleterious effects of alcohol in the liver arise as a combination of the direct effects of alcohol metabolism within the liver, and the actions and interactions of the indirect/systemic effects of alcohol with other organs and physiological systems. Following hepatic metabolism much of the hepatic damage occurs as a result of the formation of reactive metabolic intermediates that promotes hepatocyte damage and, in doing so, the activation of hepatic stellate cells, inflammation, and fibrosis. If left unchecked, progression to hepatic cirrhosis occurs, a pathological state representing the setting for approximately 80 % of all HCC diagnosed [2, 34].
Sirt1 is a histone deacetylase that is intimately positioned as a cellular metabolic switch that participates in a diverse series of biological pathways. Within the liver, Sirt1 regulates acetylation status, and thus, influences the activity of numerous transcription factors associated with lipid and glucose metabolism, including SREBP-1c, cAMP-responsive element-binding protein (CREB) regulated transcription co-activator-2 (CRTC2) and peroxisome proliferator-activated receptor gamma co-activator 1-α (PGC-1α) [35, 36]. Sustained overnutrition and alcohol consumption deplete intracellular NAD+ stores, impairing Sirt1 activity, and thus promoting lipogenesis and steatosis. Sirt1 is also attractive in targeting alcoholic liver disease because of its role in hepatic inflammation. Indeed, overexpression of Sirt1 is reported to protect against liver inflammation whereas deletion of Sirt1 enhances inflammation [37, 38]. These findings at least in part are attributed to decreased deacetylation of nuclear factor-kappaB (NF-κB) at lysine 310 [39].
In the current study, we report increases in Sirt1 expression in DEN-treated animals on a B6C3 background, but not a C57BL/6 background (Fig. 21.1b). B6C3 animals are more susceptible to liver tumor formation than C57BL/6 mice and when fed EtOH-DW, have increased HCC progression (Fig. 21.1a) [24]. However, changes in Sirt1 expression levels do not necessarily correlate to increased activity, as Sirt1 activity is more closely tied to the availability of NAD+ as a cofactor in the deacetylation reaction [40]. Thus simply having high levels of Sirt1 may be a consequence, as opposed to a cause, of increased tumor promotion in the setting of EtOH-DW for B6C3 mice.
An increasing number of reports appear to suggest a deleterious role for Sirt1 in HCC and other cancers [41, 42]. These findings are attributed to in vitro findings linking Sirt1 activity and deacetylation (and thus inactivation) of the tumor suppressor p53 [23, 43]. These findings are also coupled with observations that Sirt1 can promote cell survival through telomere maintenance [21, 44]. Despite observation findings that Sirt1 expression is enhanced in liver tumors and correlated with poor prognosis, there is a lack of in vivo data that demonstrate a mechanistic role for Sirt1 in HCC [22].
Contrary to a role in tumor promoting tumor progression, reports indicate Sirt1 impairs Wnt/β-catenin pathway-associated proliferation. Constitutive expression of Wnt/β-catenin has been found in approximately 90 % of colorectal cancers and deacetylation of β-catenin by Sirt1 has been reported to prevent transcriptional activation by β-catenin in vitro [45, 46]. Additionally, Sirt1 plays an important role in DNA stability, as evidenced by knockout of Sirt1 leading to increased genomic instability and increased chromosomal fusions [47]. Indeed, overexpression of Sirt1 prevented evidence of metabolic syndrome and provided strong protection against liver cancer development in a DEN/high-fat diet mouse model of liver cancer [48].
We present additional findings to support an anticancer role for Sirt1 activity in an in vitro model of HCC progression (Fig. 21.4). Antagonism of Sirt1 expression, utilizing the Sirt1 inhibitor EX-527, promoted HCC cell line proliferation independent of EtOH metabolism, whereas activation of Sirt1 using the specific agonist CAY10591 inhibited proliferation in both the presence and absence of 25 mM EtOH. However, it is worth noting EtOH failed to promote HCC cell line proliferation except in VL-17a cell lines expressing both ADH and CYP 2E1 (Fig. 21.4d), despite previous findings that E47 cells proliferate in the presence of an equivalent dose of EtOH [25]. Lack of a proliferative response to EtOH by VA-13 cells is not surprising considering previous findings that ADH-expressing cells have diminished proliferative responses to EtOH treatment [28].
An important consideration to the contribution of Sirt1 to the promotion or prevention of HCC is the contribution of non-parenchymal liver cells (NPCs), including Kupffer cells, sinusoidal endothelial cells, and hepatic stellate cells. Currently, there is a dearth of studies examining Sirt1 expression and/or activity on the function of NPCs; however, one report suggested Sirt1-NF-κB signaling is involved in TNF-α production in the presence of alcohol [49]. Indeed additional studies to elucidate potential role(s) for Sirt1 in NPCs utilizing models of alcohol-induced liver injury and/or cancer are attractive and may help explain differences observed in in vitro and in vivo studies presented herein.
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


