This review summarizes completed and ongoing studies evaluating the activity of immune checkpoint inhibitors in esophagogastric cancer.
Key points
- •
Immune checkpoint inhibitors that target cytotoxic T lymphocyte antigen-4 or the programmed death-1/programmed death–ligand 1 axis have transformed the treatment of many solid tumors.
- •
Initial phase I/II studies in esophagogastric cancer suggest significant activity for these drugs.
- •
Ongoing phase III studies will determine if there is a role for these drugs in the next several years.
- •
Correlative analyses are ongoing to identify the group of patients most likely to benefit from these therapies.
Introduction
Outcomes for patients with advanced esophagogastric cancer (EGC) are poor. Approximately 50% of patients with EGC present with overt metastatic disease, and chemotherapy is the mainstay of palliation in this setting. With the high likelihood that patients with initial locoregional disease will eventually have metastatic disease, palliative chemotherapy will ultimately be used in most patients. In recent years, the incorporation of targeted agents—trastuzumab with first-line chemotherapy for Her2-positive disease and ramucirumab as monotherapy or with paclitaxel chemotherapy in the second-line setting—has incrementally improved outcomes, but median overall survival (OS) remains at best only 1 year.
In this gloomy context, excitement is growing among oncologists and patients alike for the use of immunotherapy or, more specifically, immune checkpoint inhibitors. Since the landmark approval by the US Food and Drug Administration (FDA) of the anti–cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody ipilimumab in advanced melanoma, these and other antibodies (namely, antagonists of the programmed death [PD]-1/PD-ligand 1 pathway) that de-repress the immune system have undergone extensive evaluation in multiple other solid tumors, including EGC. These studies have led to the FDA approval of additional immune checkpoint inhibitors in several solid tumor malignancies and, in EGC, have culminated in ongoing phase III studies.
This review focuses on the role of the immune system in cancer, a brief history of immunotherapy, the role of immune checkpoint molecules in normal immune homeostasis and, the rapidly accumulating data in EGC.
Introduction
Outcomes for patients with advanced esophagogastric cancer (EGC) are poor. Approximately 50% of patients with EGC present with overt metastatic disease, and chemotherapy is the mainstay of palliation in this setting. With the high likelihood that patients with initial locoregional disease will eventually have metastatic disease, palliative chemotherapy will ultimately be used in most patients. In recent years, the incorporation of targeted agents—trastuzumab with first-line chemotherapy for Her2-positive disease and ramucirumab as monotherapy or with paclitaxel chemotherapy in the second-line setting—has incrementally improved outcomes, but median overall survival (OS) remains at best only 1 year.
In this gloomy context, excitement is growing among oncologists and patients alike for the use of immunotherapy or, more specifically, immune checkpoint inhibitors. Since the landmark approval by the US Food and Drug Administration (FDA) of the anti–cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody ipilimumab in advanced melanoma, these and other antibodies (namely, antagonists of the programmed death [PD]-1/PD-ligand 1 pathway) that de-repress the immune system have undergone extensive evaluation in multiple other solid tumors, including EGC. These studies have led to the FDA approval of additional immune checkpoint inhibitors in several solid tumor malignancies and, in EGC, have culminated in ongoing phase III studies.
This review focuses on the role of the immune system in cancer, a brief history of immunotherapy, the role of immune checkpoint molecules in normal immune homeostasis and, the rapidly accumulating data in EGC.
The immune system
The immune system protects us from external threats (infectious diseases) and also from internal ones (cancers) while not attacking healthy tissue (which would lead to the development of autoimmune diseases). To fulfill these critical and synchronous roles, it must recognize self from non–self-antigens with unerring accuracy.
The immune system consists of an innate and an adaptive component. The innate immune system involves rapid immune responses, which are mediated by macrophages, neutrophils, dendritic cells, and natural killer cells. These cells are hard wired to recognize non–self-antigens, such as those from infectious organisms, but have (1) relatively low potency, (2) limited specificity for the specific microorganism, and (3) no memory (ie, no ability to generate an enhanced response if re-exposed to the same microorganism).
If a microbe or cancer cell is not rapidly eliminated by innate immune mechanisms, adaptive immune responses are then engendered. These responses are produced by B cells (the humoral arm, which produces antibodies that typically target extracellular antigens) and T cells (the cellular arm, which destroys infected cells that harbor intracellular organisms or malignant cells). In contrast to innate immunity, adaptive immunity develops over days to weeks and is (1) much more potent, (2) highly specific for a specific antigen, and (3) leads to a memory response (which results in a much more rapid and potent response upon re-exposure).
Despite these coordinated mechanisms, the development of cancer necessarily implies a failure of immunosurveillance of incipient malignant cells. Dunn and colleagues proposed the concept of immunoediting to explain this phenomenon. They envisaged that this process comprises 3 phases that are collectively denoted as the 3 E s of cancer immunoediting: elimination, equilibrium, and escape. The first E refers to the fact that most cancer cells are indeed recognized and successfully killed by the immune system, leading to the second E, where the surviving cancer cells acquire multiple mechanisms that allow them to exist alongside increasingly ineffective immune responses (eg, down-regulating immunogenic molecules on the tumor cell surface or recruiting immunosuppressive mechanisms in the tumor microenvironment). This second E lasts the longest and may occur over many years. Finally, the balance of forces shifts decidedly in the favor of the cancer cells, allowing them to e scape from immune control.
Coley’s toxins
The idea of harnessing the immune system to attack cancer is an intuitively appealing concept but not a new one. The attractiveness of such a proposed treatment stems from the belief that recruitment of the immune system to attack cancer cells potentially offers more durable benefit and less toxicity than conventional therapies (akin to fighting off a virulent influenza infection) and, in some fashion, is more natural than the harsh chemicals and x-rays that comprise modern anticancer therapy.
The earliest proof of the potential of the immune system to directly combat malignant tumors stems from a series of observations and experiments by William Coley, a surgeon at the New York Cancer Hospital (the precursor to Memorial Sloan Kettering Cancer Center). He noted the regression of a soft tissue sarcoma in a patient who had erysipelas, an infection caused by Streptococcus pyogenes . He then inoculated the tumors of 10 patients directly with a culture of the bacterium and noted durable curative responses in some patients. The immune basis of these observations is not known with certainty but is presumed to involve the nonspecific recruitment to the tumor site and activation of immune cells by the bacterial products.
Although Coley’s concoction of toxins fell out of favor, other approaches to stimulate the immune system continued to be investigated in the 20th century. Most of these methods focused on vaccinating patients against preidentified antigens that are expressed preferentially or exclusively on tumor cells. Uniformly, despite laboratory evidence of cellular immune responses to these vaccines, few clinically relevant responses were noted.
The sole exception is sipuleucel-T, a recombinant human protein consisting of prostatic acid phosphatase linked to granulocyte-macrophage colony-stimulating factor, which has to be introduced into autologous peripheral blood mononuclear cells obtained from patients by leukopheresis. It was approved in 2010 for the treatment of castrate-resistant prostate cancer based on a phase III study, which found an improvement in OS in the absence of an improvement in time to progression.
The low effectiveness of most cancer vaccines is likely because of their lack of antigenicity and the failure to provide adequate costimulation (to be discussed later), which results in inactivation of T cells against the tumor.
Immune checkpoints
The generation of an effective cellular immune response by T cells against a cancer cell requires a primary signal and cosignals. As shown in Fig. 1 , the primary signal comes from recognition of a cancer-associated antigen by a T-cell receptor with high specificity for the antigen, presented in the context of a class I/II major histocompatibility complex molecule, which is expressed on so-called antigen-present cells (APCs), such as dendritic cells or on the tumor cell itself. In addition to this primary signal, a secondary signal is required, in the absence of which the T cell may be rendered anergic or nonfunctional.
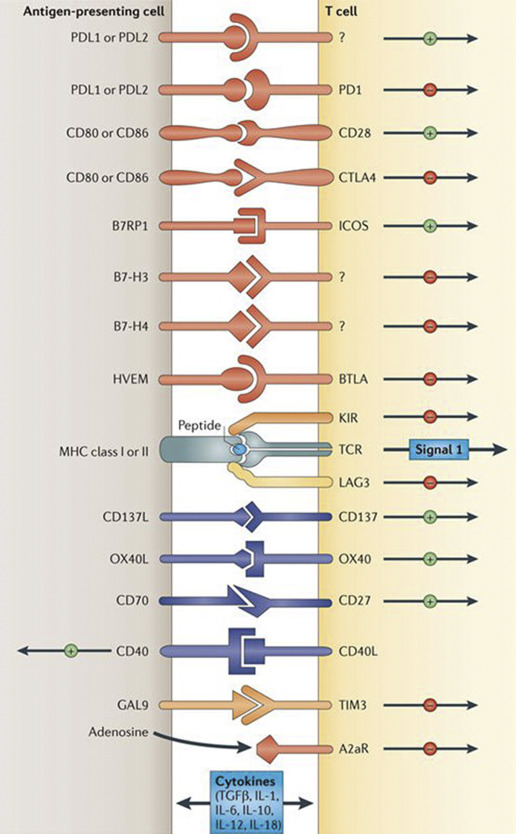
As noted in Fig. 1 , it is now well established that the interactions between multiple molecules at the interface between the T cell and tumor cell create multiple secondary signals, some of which are costimulatory and some of which are inhibitory. The ultimate activation or quiescence of the immune response depends on the complex interplay and net signal—positive or negative—of these diverse interactions, which occur at different times in the process of successful T-cell engagement and activation.
Under normal physiologic conditions, immune checkpoints are crucial for the maintenance of self-tolerance (and the avoidance of autoimmunity) and also to protect tissues from excessive damage if the immune system were to respond too exuberantly to an infection. However, many of these molecules have become co-opted by cancer cells (in the process of achieving equilibrium and escape from the immune system). Their identification and targeting with neutralizing (or agonist) antibodies now forms the basis of modern-era immunotherapy.
Cytotoxic T-Lymphocyte Antigen-4 and the Programmed Death-1/Programmed Death–ligand 1/2 Pathway
In 1995, James Allison and colleagues were 1 of 2 groups that simultaneously characterized the function of CTLA-4, a protein that has high homology with CD28, which was already known to be a costimulatory molecule expressed on T cells necessary to provide the secondary signal for T-cell activation. Just like CD28, CTLA-4 also binds their cognate ligands, the B7 molecules (which are found on APCs), but with much higher affinity. However, unlike CD28, CTLA-4 expression is induced only when a T cell becomes activated. It then competes with CD28 for binding to the B7 molecules but leads to down-regulation and eventual abrogation of the immune response.
Subsequently, PD-1 was also identified as another negative immune checkpoint molecule. PD-1 has 2 ligands, PD-L1 and PD-L2. PD-L2 is mostly expressed on APCs, whereas PD-L1 is expressed on numerous tissues, including immune and tumor cells. In the tumor microenvironment, PD-L1 expressed on tumor cells binds to PD-1 on activated T cells reaching the tumor. This delivers an inhibitory signal to those T cells, preventing them from killing target cancer cells and protecting the tumor from immune elimination. Unlike CTLA-4, which is thought to be necessary for T-cell activation, the PD-1/PD-L1/2 pathway is thought to protect cells from T-cell attack.
Anti–Cytotoxic T-Lymphocyte Antigen-4 Antibodies
The 2 anti–CTLA-4 antibodies that have been evaluated in EGC are ipilimumab and tremelimumab. In the first-line phase III study of ipilimumab in advanced melanoma, immune-related adverse events (irAEs) resulted from nonspecific immune activation and included diarrhea (33% all grade, 4% grade 3), pruritus (27% all grade, 2% grade 3), rash (22% all grade, 1.2% grade 3), and elevation in liver enzymes (about 29% all grade, 15% grade 3/4). No treatment-related deaths were reported. Since that time, the growing clinical experience with ipilimumab and other immune checkpoint inhibitors has also led to well-established algorithms for treating irAEs with the use of steroids and other immunosuppressants, which do not appear to reduce the benefit from ipilimumab in melanoma patients.
Historically, the first immune checkpoint inhibitor to be studied in EGC was tremelimumab. In a phase II study, Ralph and colleagues evaluated tremelimumab, 15 mg/kg every 90 days, in 18 patients with advanced esophageal, gastroesophageal junction (GEJ), or gastric adenocarcinoma; 15 received prior first-line chemotherapy, and 3 received prior second-line treatment. One patient achieved a partial response (PR, 6%) by standard Response Evaluation Criteria in Solid Tumors (RECIST) criteria that was ongoing at 33 months of follow-up, whereas 4 other patients achieved stable disease (SD, 22%). Although median time to progression and OS were disappointing (2.83 and 4.83 months respectively), one-third of patients were alive at 12 months. Grade ≥3 toxicities included rash and diarrhea in 2 and 3 patients, respectively, consistent with the known toxicities of these drugs. Correlative analyses included evaluating T-cell proliferative responses to carcinoembryonic antigen; the 5 patients with a posttreatment response had improved OS compared with the 8 assayed patients without a carcinoembryonic antigen response (17.1 vs 4.7 months; P = .004).
The dose of tremelimumab in this study is now considered subtherapeutic. In an ongoing study of tremelimumab (with or without the anti–PD-L1 antibody, durvalumab), the dose of tremelimumab monotherapy is 10 mg/kg every 4 weeks.
Data for ipilimumab were recently reported in abstract form. This was for a randomized phase II study in which 114 patients with either a PR or SD to first-line fluoropyrimidine/platinum chemotherapy were randomly assigned to best supportive care (BSC, which mostly consisted of continuation of the fluoropyrimidine) versus ipilimumab. The primary endpoint was immune-related progression-free survival (PFS), which used a modification of the modified World Health Organization criteria, in which the appearance of new lesions does not automatically constitute progressive disease. Unfortunately, the immune-related PFS was only 2.9 months in patients who received ipilimumab versus 4.9 months for patients who continued on fluoropyrimidine maintenance chemotherapy. The median OS was similar in both groups (12.7 vs 12.1 months). Toxicities were also higher in the ipilimumab versus BSC arm (72% vs 56%) and included pruritus (32%), diarrhea (25%), fatigue (23%), and rash (18%).
These 2 studies contain the only data for anti–CTLA-4 antibody monotherapy in EGC ( Table 1 ). They suggest modest activity for these drugs at best, and toxicity profiles comparable with the known irAEs of ipilimumab in other cancers.
| Treatment | Location/Histology | No. of Patients | Response Rate, % | PFS | OS | Reference | ||
|---|---|---|---|---|---|---|---|---|
| Median | Overall | Median | Overall | |||||
| Tremelimumab | E/GEJ/G adenoCA | 18 | 6 | 2.83 | NS | 4.83 | 33% 1-y | Ralph et al, 2010 |
| Ipilimumab | GEJ/G adenoCA | 57 | NS | 2.9 mo | NS | 12.7 | NS | Moehler et al, 2016 |
| BSC (including chemo) | 57 | NS | 4.9 mo | NS | 12.1 | NS | ||
Anti–Programmed Death-1 and Programmed Death–Ligand 1 Antibodies
Several anti–PD-1 and anti–PD-L1 antibodies are now approved for the treatment of various cancers. Pembrolizumab, an antibody against PD-1, was initially approved in 2014 for the treatment of advanced melanoma. Since then, it has also obtained approval in non–small cell lung cancer in 2015. In general, toxicities associated with pembrolizumab (and other anti–PD-1 inhibitors) seem to be less than with ipilimumab, as was noted in a phase III study in melanoma that compared 2 doses of pembrolizumab with ipilimumab and found lower rates of grade ≥3 toxicities for pembrolizumab (10%–13% vs 20%). The rates of treatment discontinuation were also lower in the pembrolizumab arms than in the ipilimumab arm (4%–6.9% vs 9.4%). Individual grade ≥3 toxicities with pembrolizumab were less than 5% and include colitis, hepatitis, and pneumonitis.
Another anti–PD-1 antibody, nivolumab, is also now FDA approved for several malignancies, including melanoma, non–small cell lung cancer, renal cell carcinoma, and Hodgkin lymphoma. Toxicities of nivolumab are qualitatively and quantitatively similar to those of pembrolizumab.
Nivolumab’s labeling indication was expanded in 2016 to permit for combination with ipilimumab as first-line therapy for advanced melanoma. This approval was based on a phase III study, which showed improvement in PFS for the combination versus ipilimumab or nivolumab alone. However, this increased efficacy is at the expense of significantly added toxicity (grade ≥3 toxicity rate of 55.0% vs 16.3% for the nivolumab arm and 27.3% for the ipilimumab arm).
An interesting observation is that, in patients whose tumors were PD-L1 negative, the addition of ipilimumab to nivolumab improved outcomes compared with nivolumab only (PFS 11.2 vs 5.3 months), whereas the addition of ipilimumab did not improve outcomes in patients with PD-L1–positive tumors (PFS, 14.0 months in both groups). This finding gives rise to the intriguing theory that PD-L1 may be upregulated by a tumor as a defense mechanism to dampen the immune system after it has been infiltrated and recognized by T cells. Therefore, in such an inflamed tumor microenvironment, PD-1 blockade alone may be sufficient to exert a significant effect. On the other hand, the absence of PD-L1 inhibition suggests a noninflamed tumor, which requires CTLA-4 blockade to drive T cells into the tumor to facilitate tumor recognition to benefit from blockade of the PD-1/PD-L1 axis.
Finally, an anti–PD-L1 antibody, atezolizumab, was recently approved to treat advanced urothelial carcinoma, based on a single-arm phase II study that treated 310 patients. Toxicities seem to be qualitatively similar to those of the anti–PD-1 antibodies and less than ipilimumab; 5% of patients had a grade ≥3 irAE, which included colitis, pneumonitis, and elevation of liver enzymes in 1% of patients each.
Although many of these anti–PD-1 and anti–PD-L1 antibodies have been evaluated in EGC (results are summarized in Table 2 ), only 1 study has been published. The KEYNOTE-012 study is a phase Ib dose-expansion study that evaluated 39 patients with GEJ/gastric adenocarcinomas, whose tumors were found to be PD-L1 positive using an experimental immunohistochemistry (IHC) assay that used the Merck 22C3 antibody. Based on the cutoff for positivity of ≥1% membrane staining of tumor or peritumoral mononuclear inflammatory cells, 40% of tumors were noted to be PD-L1 positive.
Related posts:
 Advances in Esophageal and Gastric Cancer
Gastric and Esophageal Cancer 2017
Advances in Esophageal and Gastric Cancer
Gastric and Esophageal Cancer 2017
 Minimally Invasive Surgery
The Asian Perspective on the Surgical and Adjuvant Management of Esophagogastric Cancer
Controversies and Consensus in Preoperative Therapy of Esophageal and Gastroesophageal Junction Cancers
Minimally Invasive Surgery
The Asian Perspective on the Surgical and Adjuvant Management of Esophagogastric Cancer
Controversies and Consensus in Preoperative Therapy of Esophageal and Gastroesophageal Junction Cancers
 Issues in the Management of Esophagogastric Cancer in Geriatric Patients
Issues in the Management of Esophagogastric Cancer in Geriatric Patients
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


