Fig. 3.1
Growth factor independence leads to sustained signalling in pathways that control essential biological functions. Binding of ligand to the extracellular ligand-binding domain leads to receptor dimerisation and activation of the kinase function in the intracellular domain. This mediates receptor phosphorylation, triggering recruitment of adaptor molecules and activation of downstream signal transduction pathways, leading to altered patterns of gene expression. Ultimately, this leads to altered biological behaviours which are characteristic hallmarks of cancer
Normally, activation of growth factor receptors is very tightly regulated – as is the synthesis and release of the ligands that stimulate them. Cancer cells almost ubiquitously deregulate normal growth factor signalling pathways and use them to promote unrestrained cell division [4]. It is important to note that there is no ligand for the c-erbB2/HER2 receptor and that the c-erbB3/HER3 receptor has no kinase activity. Nonetheless, these receptors are able to participate in signalling by dimerising with appropriate partners (e.g. c-erbB-2:c-erbB-3 (HER2/HER3) heterodimers exploit ligand binding to the HER3 component and kinase-mediated signalling from the HER2 component).
Cancer cells achieve self-sufficiency in growth factors by three main mechanisms: (1) they make and release growth factors to stimulate their own (autocrine) and neighbouring cells’ (paracrine) receptors; (2) they alter the number, structure or function of growth factor receptors on their surface so they are primed to relay a growth signal to the nucleus (even without binding of cognate ligand); and (3) they deregulate signalling pathways downstream of the receptor to turn it on permanently (constitutive activation). In contrast to other tumour types, in which EGFR gene amplification or mutation is frequent (e.g. lung adenocarcinoma), overexpression of the receptor, without gene amplification, is the dominant process whereby EGFR influences the pathobiology of SCCHN. Previously, a mutated, truncated form of EGFR (EGFR variant III) was believed to be expressed in many head and neck cancers [5], but recent reports have cast doubt on this as a major driver in SCCHN [6]. The roles of c-erbB-2, c-erbB-3 and c-erbB4 in SCCHN remain unclear. However, it is known that c-erbB-2:c-erbB-3 (HER2/HER3) heterodimers are potent inducers of the PI3-kinase anti-apoptotic pathway [7] and this may be relevant to particular subsets of SCCHN, including human papillomavirus (HPV)-related disease [1].
c-erbB receptors are attractive therapeutic targets in SCCHN. Two main classes of drugs, monoclonal antibodies (MAB) and small molecule tyrosine kinase inhibitors (smTKI), have been developed. MAB are large molecules directed against the extracellular domain of the receptor, while smTKI inhibit the receptor’s intracellular kinase domain. A number of EGFR-targeted MAB, differing in terms of their species of origin (murine, chimeric (part human/part murine) or fully human) and the specific part (epitope) of the EGFR protein to which they bind, have entered clinical trials. The main agents to consider in the context of SCCHN are: cetuximab (C225, Erbitux), zalutumumab and panitumumab (Vectibix). Cetuximab has been shown in randomised phase III trials to be effective in combination with radiation [8, 9] (but not chemoradiation [10]) in newly diagnosed locally-advanced disease. In addition, it has been shown to improve survival when combined with platin and 5-fluorouracil chemotherapy in the context of first-line treatment for relapsed/metastatic disease [11]. The other anti-EGFR MABs have not established a role in the treatment of SCCHN, either as part of radiation/chemoradiation (RT/CRT) combinations [12–14] or in the relapsed/metastatic setting (first- or second-line) [15, 16]. Small molecule TKIs have, as yet, failed to find a place as standards-of-care in the management of SCCHN. Gefitinib, erlotinib and lapatinib have been evaluated in combination with RT/CRT in patients with newly diagnosed locally-advanced SCCHN and in relapsed/metastatic disease, without clear evidence of benefit [17–26]. In the context of relapsed/metastatic disease, only one agent (afatinib) has shown a positive result in a randomised trial [27]. Afatinib is an irreversible inhibitor of EGFR, c-erbB2/HER2 and c-erbB4/HER4 that is currently being investigated in a number of settings in SCCHN. In the primary treatment setting, it is being tested in combination with CRT (NCT01732640), and as adjuvant therapy after definitive CRT in high-risk patients (phase III LUX2 study NCT01345669) or after surgery (GORTEC 2010–02, EudraCT 2010-023265-22). Afatinib has been assessed in the phase III LUX head and neck-1 study in 583 patients receiving second-line therapy for relapsed/metastatic SCCHN [27]. Afatinib significantly increased median progression-free survival (2.6 versus 1.7 months, p = 0.03) but did not improve median overall survival (6.8 versus 6.0 months) relative to methotrexate. In an integrated analysis of quality of life, afatinib showed a delay in deterioration of global health status, pain and swallowing problems (all p≤0.03). These modest benefits mean that it is unlikely that afatinib will be approved for use in patients with relapsed/metastatic SCCHN.
Toleration of DNA Damage and Avoidance of Cell Death
During cell division, as cells duplicate their DNA, they assess their genomic integrity and check for errors in the DNA sequence. If errors are present, they can arrest at cell cycle checkpoints and engage DNA repair pathways. There are four cell cycle checkpoints: at G1/S transition, during S phase, early in G2 phase and late in G2 phase.
Two of these checkpoints are particularly relevant to the cellular response to RT/CRT in SCCHN [28]. The G1/S checkpoint allows diploid (2n, 46 chromosomes) cells to pause before entering synthesis (S) phase, during which the entire DNA complement is duplicated to create a cell with 4n DNA content before mitosis. The tumour suppressor genes TP53 and retinoblastoma (RB1) are essential for the G1/S checkpoint. TP53 pathway mutation or inactivation is very common in SCCHN. Smoking-induced SCCHN shows almost universal loss-of-function mutations of the p53 pathway, with 84 % of tumours showing TP53 mutation [1]. In contrast, just 3 % of HPV-negative SCCHNs have mutated TP53, but in these tumours the pathway is inactivated through interactions between viral E6 and E7 proteins with p53 and Rb proteins, respectively. As a consequence of aberrant G1/S checkpoint status, most SCCHN are highly reliant on a functional G2/M checkpoint.
RT/CRT induces a number of lesions in DNA, ranging from purine and pyrimidine lesions to single-strand (SSB) and double-strand breaks (DSB) in the DNA. DSB are the most lethal DNA lesions induced by RT/CRT and therapies that can prevent their repair/resolution have the potential to act as radiosensitisers. As might be expected, there are specific mechanisms to detect and repair radiation-induced abnormalities in DNA structure: DSBs are repaired by non-homologous end-joining (NHEJ) repair during G1 phase of the cell cycle and by high-fidelity homologous recombination (HR) in S and G2 phases; SSBs and base damage are repaired through the base excision repair pathway (reviewed in 28).
Normal (and cancer) cells continually assess their viability by auditing the balance of pro- and anti-survival signals that they receive. In normal cells, endogenous (during replication) or exogenous (from genotoxic events such as RT/CRT) DNA damage leads to cell cycle arrest and attempted DNA repair. If the extent of damage exceeds the capacity for repair, the balance of pro- and anti-survival signals shifts and the cell triggers cell death pathways (apoptosis, autophagy, necroptosis, necrosis). This mechanism protects the organism against generating and perpetuating genetic damage. In contrast, loss of normal cell death pathway signalling is extremely common in cancer (including SCCHN). Indeed, two of the best known cancer-associated genes (TP53 and BCL–2) are intimately involved in cell death signalling. Cancer cells frequently evade apoptosis by ignoring signals sent through the extrinsic pathway or by resetting the balance of intracellular pro- and anti-survival molecules in favour of inhibition of apoptosis. By avoiding apoptosis, cancer cells can sustain DNA damage without it causing cell death (unless the damaged gene is absolutely necessary for cell survival). Thus, cancer cells that bypass normal apoptotic signalling are more likely to have unstable genomes and to be intrinsically resistant to anti-cancer treatments.
Although there are obvious advantages in cancer cells switching off normal cell cycle checkpoint controls and cell death signalling pathways, it also exposes them to significant risks. As discussed above, most SCCHN (both HPV-negative and HPV-positive) lack a normal p53-mediated G1/S checkpoint and, thus, become highly dependent on the ATR-Chk1 pathway (Fig. 3.2) to mediate S and G2/M arrest to allow repair of DNA damage following RT/CRT. As such, G2/M checkpoint control can be seen as a particularly attractive target for cancer-specific radiosensitisation [reviewed in 28]. p53-defective cancer cells exposed to RT/CRT will not be able to arrest at the G1/S checkpoint, rendering them highly vulnerable to drugs that inhibit the G2 checkpoint. Cancer cells treated in this way will proceed into mitosis before repairing their RT/CRT-induced DNA damage and, as a result, will run a significant risk of mitotic catastrophe and cell death. This activity of G2/M checkpoint inhibitors in p53-defective tumour cells is an example of so-called synthetic lethality and represents a promising prospect for developing cancer-selective radiosensitising drugs (Fig. 3.3).
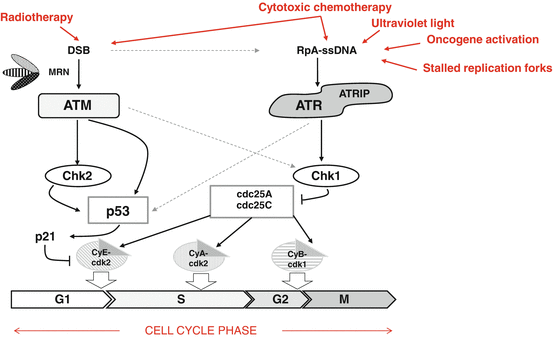
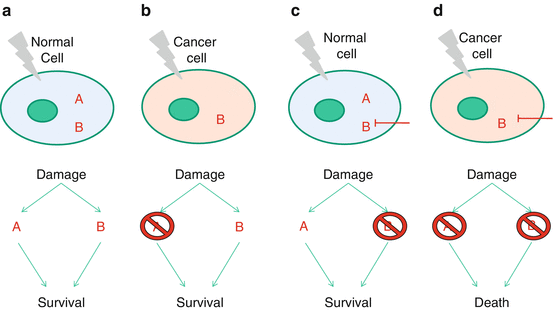
Fig. 3.2
DNA damage leads to activation of DNA damage response and cell cycle arrest pathways. The frequent loss of p53 function in head and neck cancers leads to reliance on G2/M cell cycle arrest and DNA repair pathways that operate in G2 phase of the cell cycle
Fig. 3.3
The concept of synthetic lethality as applied to G2/M-targeted drugs in tumours that lack p53-mediated G1 checkpoint control
A number of G2/M-targeted radiosensitisers are under development and are beginning to enter clinical trials. ATR inhibitors (VE-821, VE-822, AZD6738) are showing promise in preclinical models and are in early-phase clinical trials both as monotherapy and combined with radiotherapy, chemotherapy or immunotherapy [NCT02223923, NCT02264678] [29–33]. Early Chk1 inhibitors (UCN-01, AZD7762) have been tested in clinical trials, but their development was stopped because of concerns about lack of efficacy or toxicity [34–38]. However, newer orally bioavailable agents, such as CCT245737 [39], are now in clinical trials and would appear to offer significant promise as targeted chemo-/radiosensitisers.
Invasion and Metastasis
Distant metastases cause 90 % of cancer deaths and are emerging as an increasing problem in SCCHN as we improve our ability to secure locoregional control with advanced RT/CRT and surgical approaches. Invasion and metastasis involves orchestrating a sequence of complex biological processes [40]. These include: (1) detachment of the tumour cell from its immediate neighbours and stroma at the local site; (2) enzymatic degradation of the extracellular matrix followed by specific directional motility (as single cells or in co-operative groups); (3) penetration (intravasation) of blood or lymphatic vessels and tumour embolisation; (4) survival of tumour cells in the circulation until arrival at the metastatic site that may be “chosen” on the basis of its ability to provide a favourable supply of appropriate growth factors; (5) attachment to the endothelium of blood vessels at its destination and extravasation from the vessel; and (6) proliferation and invasion of its new location and recruitment of a new blood supply.
This general scheme has been understood for some years, but a number of recent observations have shed new light on the process of invasion and metastasis. In an in vitro culture system that attempted to recapitulate the physiological situation (surface exposed to a gaseous interface, base in contact with a matrix of fibrillar collagen I, laminin, collagen IV), Gaggioli et al. (2007) [41] showed that the movement of malignant head and neck cancer cells through the extracellular matrix was a collective process that enlisted the help of cancer-associated fibroblasts. Using imaging and molecular biology techniques, it was shown that fibroblasts generate tunnels in the extracellular matrix, by enzymatic (protease) digestion and force-mediated matrix remodelling, through which carcinoma cells follow. The carcinoma cells use Cdc42 and MRCK (myotonic dystrophy kinase-related CDC42-binding protein kinases) to regulate intracellular myosin light chain as a means of controlling their directional motility behind invading fibroblasts. Some of these signaling events are controlled by proteins which may be druggable targets, offering the prospect of developing specific agents that inhibit the ability of malignant cells to invade normal tissues. Such studies are ongoing.
The process by which tumour cells escape from the primary tumour and establish metastatic colonies in biologically favourable secondary tissues has also been the subject of much research. This work has completely changed our understanding of the biology of tumour metastasis. For example, studies have shown that far from being a passive game of chance whereby an aspiring metastatic cell has to trust fortune to carry it to what Paget called “congenial soil”, it is now clear that metastasis involves a degree of forward planning and preparation on the part of the tumour. In breast cancer models, it has been shown that metastasis is facilitated by the formation of “pre-metastatic niches” in target organs. This occurs through the activities of bone marrow-derived cells that accumulate at metastatic sites in advance of tumour deposits and condition the environment to make it favourable to tumour cells that subsequently arrive and establish a secondary colony. The process is initially triggered by hypoxic tumour cells secreting an enzyme, lysyl oxidase (LOX), into the circulation. LOX accumulates at pre-metastatic sites where it mediates the cross-linking of structural proteins in the extracellular matrix [42]. Specifically, LOX catalyses oxidative deamination of the primary amines of lysine and hydroxylysine in proteins such as collagen and tropoelastin to generate peptidyl-α-aminoadipic-δ-semialdehyde, an aldehyde that spontaneously condenses to form both inter- and intra-chain cross-links. Consequently, LOX regulates maturation of proteins in the extracellular matrix, thereby modifying its tensile strength and function and playing an important part in connective tissue remodelling. LOX-mediated tissue remodelling is an essential prerequisite for subsequent recruitment of CD11b + myeloid cells which adhere to crosslinked collagen IV and produce matrix metalloproteinase-2, which cleaves collagen and increases the invasion and recruitment of bone marrow-derived cells and metastasising tumour cells. Elevated LOX expression is associated with the formation of metastases but it is also required for their continued survival and growth. Importantly, it has been shown that inhibiting LOX can prevent CD11b + cell recruitment and, thus, prevent metastatic growth. Le et al. (2009) have validated LOX as a biomarker of metastasis in head and neck cancer patients [43]. They studied 66 head and neck cancer patients from a single institution and 306 of 1113 patients treated in the RTOG 90–03 trial. LOX expression predicted time to metastasis in the initial 66 patient cohort. In the RTOG 90–03 study, multivariate analysis, controlling for significant parameters such as nodal stage and performance status, revealed LOX expression was a statistically significant independent predictor for time to metastasis, time to progression and overall survival. These fundamental discoveries have resulted in drug discovery programmes that seek to generate chemical inhibitors of LOX activity, with the goal of reducing the establishment of metastatic deposits and attacking those that have already come into being.
Evading Immune Destruction
The theory of immune surveillance maintains that the immune system is constantly vigilant against the emergence of premalignant and frankly malignant cells. The observation that patients with chronic immunosuppression (e.g. after organ transplantation) are prone to specific cancers, especially those of viral origin, is frequently cited as evidence in favour of this theory. There is also evidence that the immune system acts as a significant barrier to non-virally-induced cancers in immunocompetent patients. Therefore, the occurrence of tumours can be viewed as a failure of the immune system to recognise, reject and destroy tumour cells that express altered self-antigens. As part of this process, it is thought that selection of less immunogenic cancer cells (through immunoediting) and active recruitment of immunosuppressive components of the immune system (e.g. regulatory T cells, myeloid-derived suppressor cells and suppressive macrophages) to some cancers ultimately allows tumours to escape from immune surveillance, grow and spread without becoming targets for immune attack [44, 45].
For decades, scientists and clinicians sought to understand how they could use immunotherapies as active anti-cancer agents. Much of this work focussed on using immune-activating cytokines (e.g. interleukin-2, interferon-α, interferon-β) as single-agent treatments or as part of biochemotherapy cocktails. In the main, those efforts proved to be fruitless. It is only in recent years that immunotherapy has emerged as a major new modality in the treatment of a wide variety of solid cancers, including SCCHN [44]. Renewed interest in immunotherapy has been underpinned by significant advances in our understanding of the fundamental biological principles that govern the activity of the immune system. In particular, specific immune checkpoints have been discovered that act as integral components of normal immune responses. In normal health, these checkpoints function to inhibit T cells and prevent them from becoming chronically activated or aberrantly directed against normal tissues. Effectively, they serve the function of negative regulators or “brakes” on the normal immune response. Many cancers exploit these inhibitory pathways in order to break free from immunosurveillance. Therefore, by activating brakes on the immune system, cancer cells are able to emerge from elimination/equilibrium phase and escape from immune detection and/or immune-related attack [45].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


