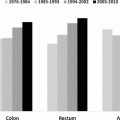
Fig. 3.1
Incidence of invasive cancer of the colon, rectum, and anus, SEER18, 2000–2012 by age, site, and sex. Age groups with <10 patients excluded. *Includes rectosigmoid junction
AYA CRC is known to exhibit a greater frequency of mucinous histology, the presence of signet ring cells, high MSI, and a higher incidence of mutations in mismatch repair (MMR) genes [3–5, 29–31]. Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominant syndrome that is associated with an approximately 70 % lifetime risk of CRC (often right sided) and a 50–70 % risk of endometrial cancer [32]. It is caused by heterozygous mutations in one of four MMR genes MSH2, MLH1, MSH6, or PMS2 (predominantly MLH1 and MSH2) and is associated with colon cancer that can appear in patients as young as their mid-20s, as well as in older adults. Silencing of the MLH1 gene by methylation is observed in 20 % of sporadic CRC. Colorectal tumors with MLH1 silencing or from patients with HNPCC exhibit MSI. While some cases of AYA CRC exhibit MSI due to a hereditary component, many do not have a history of genetic predisposition, suggesting that de novo somatic mutations in MMR genes may be a molecular feature of AYA CRC. Relatively few molecular genetic studies have been conducted in this age group, perhaps due to the fact that these cases are few in number and tissue samples are difficult to procure. Recent work by the Cancer Genome Atlas (TCGA) study has provided data on genes that are frequently mutated in adult CRC [33]. This and other studies have identified several genes that exhibit amplification and elevated expression in adult CRC, including IGF-2 [34]. There are also consensus gene sets exhibiting mutations in adult CRC that have been identified [35, 36]. These data provide a baseline for pathway analysis that could direct us toward novel signaling pathways in AYA CRC tumors. While AYA CRC tends to have a more mucinous phenotype and appears to have a higher frequency of MSI even in the absence of a hereditary component, molecular targets that would make the AYA CRC more vulnerable to a specific therapeutic target have yet to be identified. The only molecular marker at this time, which could be considered a predictor in AYA CRC, is MSI. However, this falls more into the category of descriptor than predictor, as does the mucinous histology phenotype. As true diagnostic markers that are specific to AYA CRC have yet to be identified, there is a need for both basic biological and translational research studies to elucidate any fundamental differences between adult and AYA CRC. The biological questions include whether there are signaling pathway differences between the two types of CRC and, if so, how unique are they to each form of the disease and how dependent are the tumor cells on these pathways for their proliferation and survival. High-throughput methodologies for gene expression and mutational analysis are expected to provide initial clues as to whether there are unique molecular characteristics associated with AYA CRC compared to that found in adults. These studies could include microarray analysis for mRNA and miRNA expression signatures and whole-exome sequencing studies for identification of unique somatic mutation patterns. Studies could also include single-nucleotide polymorphisms, methylation, and proteomic analyses to compare adult and AYA forms of the disease. The results from these studies will provide a foundation from which novel diagnostic, prognostic, and predictive markers can be identified in the AYA CRC population and perhaps reveal preferred signaling pathways that can be used as targets for therapy.
3.3.2 Acute Lymphoblastic Leukemia
As mentioned in the “Introduction,” ALL is another cancer in which there are significant data supporting a biological uniqueness to the disease in AYA patients. ALL is the most common cancer in individuals from birth to 21 years of age and is one of the leading causes of cancer-related mortality in AYAs [13, 37–39]. While the incidence of ALL in the USA, in children from 1 to 10 years of age, has remained relatively constant over the past several decades, data from the NCI SEER program reveal a continual increase in the incidence of ALL in the AYA population (ranging in age from 15 to 29 years) from 1975 to 2010 (Fig. 7.5, Chap. 7). Unfortunately, in contrast to pediatric ALL in which scientific and clinical advances have led to impressive improvements in overall survival with more than 80 % of children (depending on race, ethnicity, and age) now achieving cure on contemporary chemotherapy regimens, overall survival in AYA ALL remains poor. Less than 45 % of AYA ALL patients currently achieve long-term remissions. While ALL is one of several cancers with a poor outcome in the AYA population, ranked as 4th among the AYA cancers with the lowest overall survival rates [40], it remains the most common cause of AYA cancer-related mortality due to its high incidence in this age group [41]. The impact of age on relapse-free and overall survival in ALL is striking [17]. In addition, the annual average percent change in the death rate during 1998–2011 shows the least improvement in the AYA age range (25–44 years) compared to improvements in children and in older adults (Fig. 7.9, Chap. 7).
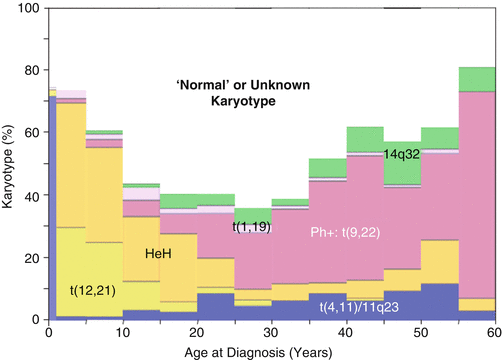
The reasons for the differences in outcome observed in pediatric vs. AYA ALL patients are due to distinct genetic and biologic features of the disease at different ages, differences in therapeutic approach and therapeutic intensity, possible differences in compliance with therapy at different ages, and/or other social and behavioral factors and are now under intensive investigation. The identification of genomic lesions has provided critical insights into leukemogenesis and is central to accurate diagnosis and current methods of risk stratification for pediatric ALL patients participating in clinical trials. Several of these genetic alterations are typically associated with a very high risk of treatment failure and relapse on current treatment regimens, the most important of which are the Philadelphia-like mutations that are analogous to Ph-1 (BCR–ABL1) but are different at the molecular level (CRLF2, JAK–1, etc.). The Ph-like mutations have a peak incidence during the AYA years (Fig. 3.2) [42, 43]. Other mutations that are more common in AYAs than children include MLL rearrangements and BCR–ABL1 itself. Importantly, as shown in Fig. 3.3, the frequency of genomic rearrangements (e.g., ETV6–RUNX1– t(12;21)) and hyperploidy associated with a superior outcome in pediatric ALL decrease in AYA ALL patients. Conversely, those genetic abnormalities associated with a poorer outcome (BCR–ABL1– t(9;22), MLL rearrangements) increase [20]. It has been found that a substantial number of children with ALL, in particular those who are older than 10 years of age, including many who relapse, lack one of these well-known chromosomal alterations associated with better outcome. This suggests that genomic/biological differences in AYA ALL may contribute to a more aggressive and treatment-refractory form of the disease.
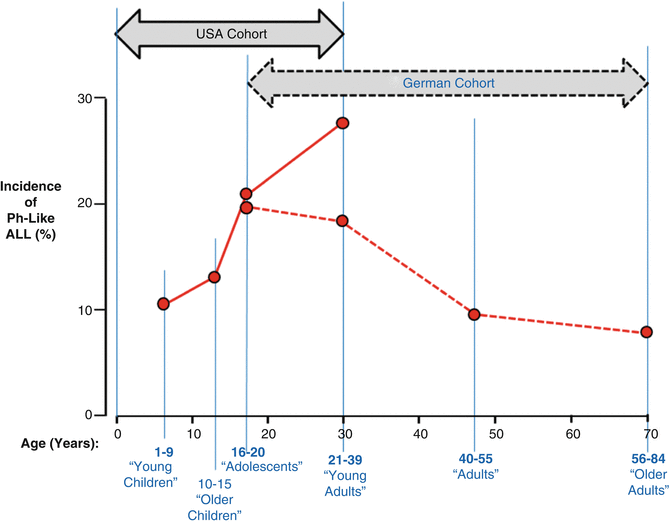
Fig. 3.2
Proportion of all pre-B-ALL that is Ph-like by age at diagnosis. The German cohort was composed of patients who received treatment on the German Multicenter Study Group for Adult ALL trials [42] and the US cohort was composed of the patients of the Children’s Oncology Group and St. Jude Children’s Research Hospital [43]
Ongoing studies in ALL xenograft/primagraft models and in human patients have demonstrated that some of the gene fusions in Ph-like ALL involving tyrosine kinases are particularly sensitive to various tyrosine kinase inhibitors (TKIs), such as imatinib or dasatinib [20, 44–46]. These observations underscore the importance of rapid translation of these genomic studies to prospective identification of patients with Ph-like ALL and therapeutic targeting of these patients with TKIs in the context of clinical trials. The identification of these patients at diagnosis will provide an opportunity to incorporate TKI inhibitor treatment to current chemotherapeutic regimens that for some has already prolonged survival [43] and as such exemplifies how a better understanding of the genomics and biology of AYA cancer can lead to better diagnostics and treatment.
The discovery of RAS mutations in a distinct cohort of 10 % of AYA ALL patients [48] also underscores the importance of developing effective treatments targeted to activated RAS and opens up avenues of investigation into the contribution of RAS pathways [20, 47–49]. Such studies will provide the mutational profiles and associated pathways that may distinguish AYA ALL from pediatric and adult forms of the disease.
3.3.3 Breast Cancer
Breast cancer is the most frequent form of cancer in women in the AYA age group throughout the world [2, 50–52, Chap. 2]. In recent years, the proportion of women presenting with distant involvement, which is associated with a poorer prognosis, has shown a statistically significant increase [53]. Age at diagnosis is an independent factor in prognosis, and cumulative evidence has suggested, but not demonstrated, that AYA breast cancer exhibits differences in type, grade, and aggressiveness in comparison to the disease in older women [2, 54]. Young women have nearly twice the rate of the basal type of breast cancer than that of any other age group (Fig. 3.4) and a peak incidence of HER–2–NEU-positive and triple-negative subtypes (Fig. 3.5) [8]. Ductal carcinoma in situ (DCIS) is 3–5× more likely to be either triple negative or HER–2–NEU positive in 25- to 30-year-olds than at any other age [8].
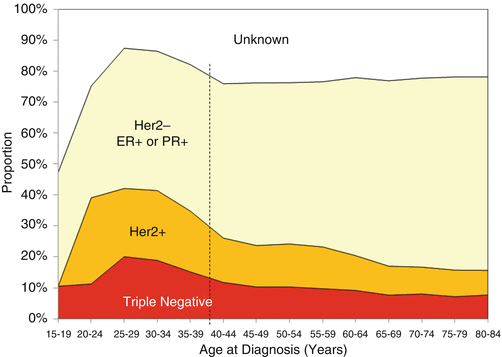
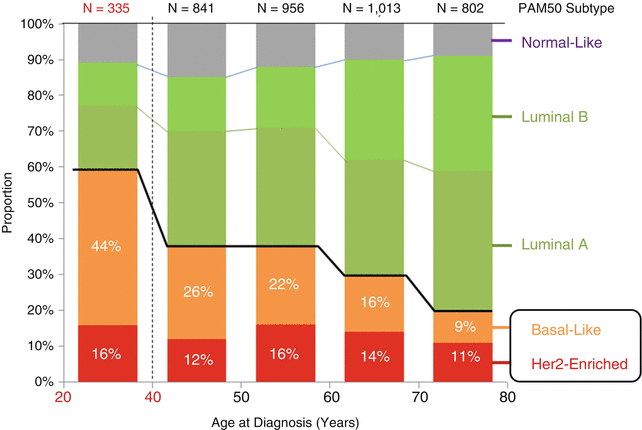
Fig. 3.5
Incidence of invasive breast cancer and ductal carcinoma in situ, SEER18, 2010–2011, by surface receptor type and age [8]
However, the hypothesis that this is due to a unique biology continues to be debated [7]. Current studies utilize gene expression profile comparisons in order to identify specific genes and molecular profiles that could identify unique factors involved in younger women with breast cancer [55–59]. The results have suggested that there are some detectable expression pattern differences between tumors in young and older women, but reanalysis of these studies has led to questions about some of the earlier conclusions [51]. Ongoing studies have focused on specific genes such as BRCA1, TP53, and others that have been linked to the early incidence of aggressive breast cancer [60, 61]. There is also a growing interest in the role of an apparent difference in tumor-associated stroma in AYA breast cancer [62]. Studies of the molecular mechanisms of different breast cancer subgroups identified in adults are likely to be informative for AYA breast cancer as well, since no consistently unique molecular or biological factors have been identified definitively [63]. Detailed studies of triple-negative/basal subgroups [64] are particularly relevant since these have been associated more frequently with breast cancers in the AYA population. This is illustrated in Fig. 3.5 showing the relative proportions of invasive breast carcinomas and DCIS as they relate to Herceptin, estrogen receptor, and progesterone receptor status. Studies on the role of the microenvironment/stroma in tumor initiation and progression will be informative for studies of AYA tumors. More extensive, carefully controlled analysis of gene expression signatures in AYA tumors relative to the same subtypes in older patients is needed to determine definitively whether a specific pattern is linked to AYA breast cancers [65]. Whole-genome analysis with deep sequencing could also help to identify mutations or polymorphic patterns that could be linked to susceptibility to early-onset breast cancer [66, 67].
3.3.4 Melanoma
The incidence rate of melanoma in children and adolescents is rising yearly and increases with age. Melanoma accounts for <1 % of all malignancies in patients less than 15 years of age compared with 8 % among AYAs and a peak of 13 % between 25 and 30 years of age. In AYAs, the incidence is higher in females than in males, whereas in older adults, melanoma is far more frequent in males (Fig. 10.1 in Chap. 10). The etiology of melanoma in the AYA population is not clear, and while diagnosis at an older age is linked to lifelong exposure in genetically less susceptible individuals, in AYAs it is likely that melanomas result from genetic and/or environmental factor interactions involving excessive UV exposure among susceptible individuals. Known predisposing factors in pediatric melanoma include rare conditions such as large congenital nevi, giant hairy nevi, acquired dysplastic nevus syndrome, xeroderma pigmentosum (XP), Werner syndrome, retinoblastoma, immunosuppression, familial melanoma syndrome, and, to a lesser extent, exposure to UV radiation [22]. How this may relate to the biology of AYA melanoma is unclear. According to staging criteria for adult melanoma, important prognostic criteria are thickness of the primary tumor, presence or absence of ulceration, nodal status, and presence or absence of metastasis. The 2007 guidelines also include using mitotic rate as a prognostic factor in staging, particularly for lesions <1 mm thick (T1) [68]. To date, however, there has been no validated staging system for melanoma in the children and AYAs. Diagnosis of melanoma in children and AYAs can be difficult since many pediatric skin lesions (such as pyogenic granulomas, Spitz nevi, and benign nevi) share some features of adult melanomas.
Molecular techniques such as comparative genomic hybridization (CGH), array-CGH, FISH, PCR-based loss of heterozygosity studies, and immunohistochemical markers have all been applied to formalin-fixed, paraffin-embedded archived clinical specimens. These approaches promise more accurate classification to discriminate a nevus from melanoma, especially in the cases of melanocytic tumors with ambiguous histology [69]. In particular, CGH may be helpful when routine histologic examination does not permit distinguishing melanoma from these lesions. However, these approaches have not been validated in children and AYAs.
Although contradictory data have been reported regarding outcome in pediatric patients [23], prognosis in the pediatric and AYA groups is generally believed to be similar to that in adults. Melanomas appear to be thicker in young patients, and metastases to sentinel nodes (SNL) are found more frequently in children and AYAs with melanoma than would be expected in adults with the same stage of disease. High mitotic rate and younger age are considered to be predictors of SLN positivity [24]. Interestingly, melanoma in young patients is also less likely to recur in distant organs. This implies that melanoma in children and AYA patients display biological differences compared with adult melanoma. For example, melanomas in young patients are more prone to progression and subsequent metastasis than in adults [25].
Marked differences exist between the patterns of melanomas and melanocytic nevi, such that neoplasms of Spitzoid morphology commonly seen in younger patients and the genetic differences between melanoma and benign Spitzoid neoplasms identified by CGH may be used to distinguish these lesions when pathological features are in question. Spitz nevus, blue nevi, proliferating congenital nevi, or other benign melanocytic lesions are characterized by fewer or absence of chromosomal aberrations or have a restricted set of aberrations with no overlap to melanoma. More information is known about genomic differences between pediatric and adult melanoma than for AYA melanoma. Some chromosomal and genomic alterations differentiate between adult and AYA melanoma. For example, frequent deletions of chromosomes 9p (the most common aberration in melanoma harboring CDKN2A), 1p, 6q, 10q, and 8p and gains of chromosomes 7, 8, 6p, and 1q are present in adult melanoma cases. Recurrent Spitz nevi revealed higher frequencies of 11p gain implicating HRAS as a candidate oncogene on 11p [70]. Pediatric melanomas demonstrate higher levels of MSI than melanomas in adults [71].
Melanoma development has been linked to germline mutations in genes encoding CDKN2A, CDK4, and MCIR and to somatic mutations in proto-oncogenes BRAF, NRAS, and KIT and tumor suppressor genes CDKN2A, TP53, and PTEN [72]. A recent study revealed that BRAF mutations that are common in melanocytic nevi and melanomas were not detected in typical Spitz nevi. Somatic mutations in BRAF are the most common genetic alterations, occurring in up to 66 % of adult malignant melanomas and 21 % of common benign melanocytic nevi with non-Spitzoid morphology [73, 74]. The most common BRAF V600E mutation occurred more frequently among patients who were 50 years of age or younger when compared with those over age 50 years. Similarly to BRAF, NRAS is mutated more frequently in melanomas than Spitz nevi, whereas no HRAS mutations have been found in Spitzoid melanomas. Approximately 67 % of Spitz nevi with increased copy number of chromosome 11p harbor an activating mutation of this oncogene that is part of RAS/RAF/MEK/ERK kinase signaling pathway. This is in contrast with rare HRAS mutations in Spitz nevi with normal HRAS copy number.
Pediatric melanomas demonstrate more frequent loss of INK4A and gain of KIT oncogenes compared with familial melanoma cases. Gain of KIT may be associated with metastasis of pediatric melanoma [75]. Early development of melanoma in children and AYAs occurs following loss of key regulators of melanocyte function. Currently, the p16INK4 tumor suppressor gene on chromosome 9p21 is postulated as a strong candidate melanoma gene, with particular relevance to early-onset familial melanoma. Spitz nevi, as a group, show high levels of p16 expression [76]. Whether sporadic melanomas occurring in childhood and AYAs are associated with aberration of this gene needs to be confirmed. In addition, expression of cyclins D1, D3 A, and CDK4 in adult and young adult/pediatric melanomas should be explored for comparison of melanoma specimens across age groups.
Recent findings suggest that the differential biology of melanoma at different ages is driven partly by deregulation of microRNA (miRNA) expression that target proliferation genes including MAP3K5, RAS-related protein RAB32, and the suppressor of cytokine signaling (SOC1), inflammation (cytokines and chemokines) EMT transition, and stroma (disintegrin and metalloproteinase precursors and TNF-related proteins) [77]. Several miRNA species have been identified to be either up- or downregulated in melanomas from different age groups. miRNA profiles in the primary melanomas may be associated with the clinical parameters of stage and nodal involvement that differ in adult and young populations, suggesting that melanomas in older adults rely on different pathways of invasion than young adult melanomas. The outcome for younger patients may be improved, but stratification for pediatric and AYA age groups should occur in biology studies and clinical trials so that prospective data are obtained for this population in which the incidence of melanoma is rising. Improved knowledge of the biology of tumors in this population and formal testing of novel-targeted therapies and immunotherapy approaches are required to improve outcomes of melanoma in AYA patients.
3.3.5 Sarcoma
In AYA patients, sarcomas represent 5.3 % of all invasive cancers, with more sarcomas in the age group located in soft tissue (3.9 %) than in bone (1.3 %) (SEER18, 2000–2012) [8]. Derived from the mesenchyme of connective tissues, they are the most diverse and heterogeneous of cancers grouped by standard classification systems. The most common sarcomas in AYAs, in descending order of incidence according to the US SEER database for 2000–2012, are Kaposi sarcoma, dermatofibrosarcoma protuberans, leiomyosarcoma, osteosarcoma, peripheral primitive neuroectodermal tumor (PNET) (originally called Ewing sarcoma), myxoid chondrosarcoma, myxoid liposarcoma, gastrointestinal stromal tumor (GIST), fibrous histiocytoma, synovial sarcoma, phyllodes tumor, spindle cell sarcoma, fibromyxosarcoma, alveolar rhabdomyosarcoma (ARMS), hemangiosarcoma, epithelioid sarcoma, desmoplastic small round-cell tumor (DSRCT), embryonal rhabdomyosarcoma, fibrosarcoma, liposarcoma, angiomatoid fibrous histiocytoma, alveolar soft part sarcoma, inflammatory myofibroblastic tumor, and giant cell tumor of bone [8]. The incidence of each of these types of sarcoma with respect to age varies with diagnosis (Figs. 3.6 and 3.7).
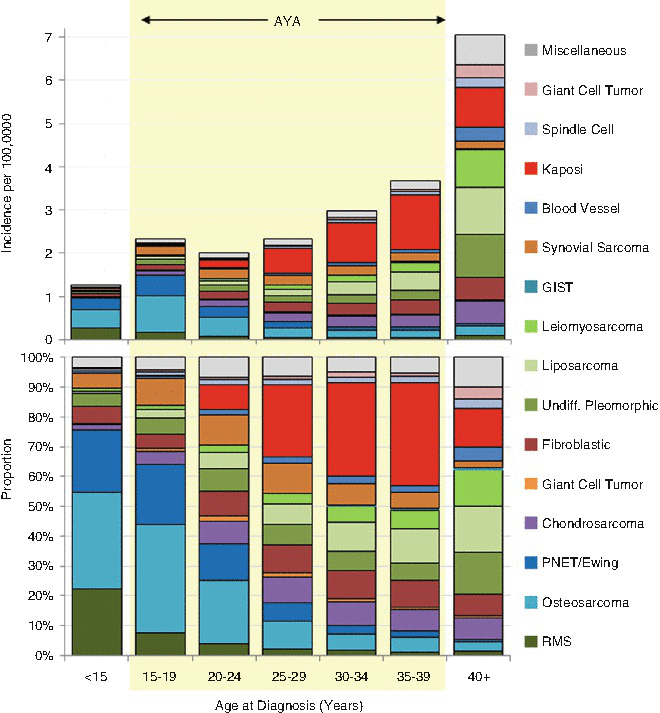
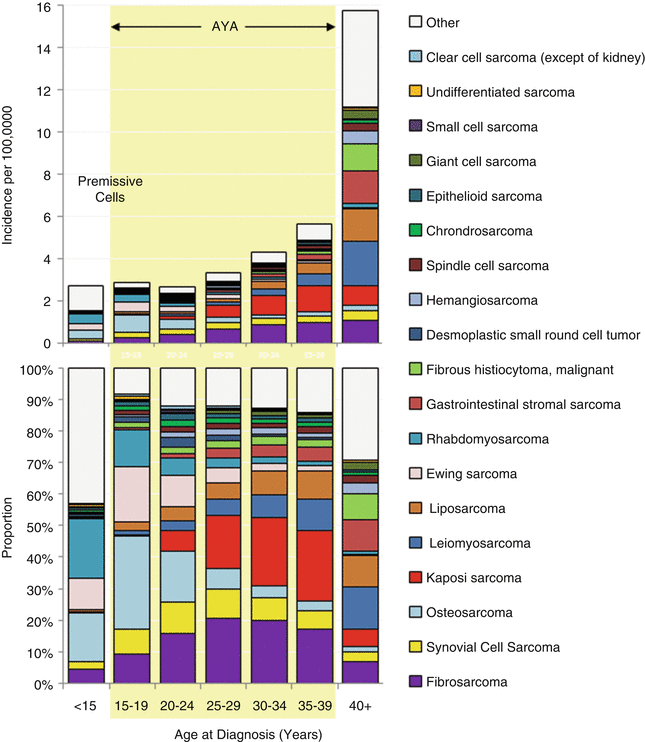
Fig. 3.6
Incidence and distribution by ICD-O-3 histology code of soft-tissue and bone sarcomas, US SEER18, 2000–2012, by 5-year age intervals in AYAs and younger and older patients. GIST-gastrointestinal stromal tumor
Fig. 3.7
Incidence and distribution by ICD-O-3 histology code of soft-tissue and bone sarcomas, US SEER18, 2000–2012, by 5-year age intervals in AYAs and younger and older patients. Sarcoma classification differs from Fig. 3.6
More than 50 morphological subtypes of sarcoma exist that in 2013 were recategorized by the WHO Classification of Tumors of Soft Tissue and Bone [78]. This revision was driven primarily by the rapidly increasing knowledge of the genetics of sarcomas, two aspects of which are particularly important for AYA patients. First, this classification is more reproducible and provides a universal nomenclature that helps to ensure comparability of international trials, facilitation of translational research, and ultimately more effective treatment. In addition, more than 60 hereditary syndromes related to bone and soft-tissue sarcomas have been recognized in this volume [78], most of which are detectable early in life and allow earlier diagnosis of the related malignancy and some of which primarily affect AYAs. In addition, many of these sarcomas have been identified to have gene fusions (Table 3.1).
Table 3.1
Associated gene fusions in sarcoma
Sarcoma type | Cytogenetics | Fusion genes | Gene | Frequency |
|---|---|---|---|---|
Angiomatoid fibrous histiocytoma | t(2;16)(q34;p11) | FUS-CREB1 | bZIP | 89 % |
t(12;16)(q13;p11) | FUS-ATF1 | bZIP | 11 % | |
Alveolar soft part sarcoma | t(X;17)(p11;q25) | ASPSCR1- TFE3 | Microphthalmia-TFE, basic helix-loop helix, leucine zipper | 100 % |
Alveolar rhabdomyosarcoma | t(2;2)(q35;p23) | PAX3-NCOA1 | Paired box/homeodomain | Rare |
t(2;13)(q35;q14) | PAX3-FKHR | Paired box/homeodomain | 95 % | |
t(1;13)(p36;q14) | PAX7-FKHR | Paired box/homeodomain | 5 % | |
Ewing sarcoma | t(11;22)(q24;q12) | EWSR1-FLI1 | Ets-like | 85 % |
t(21;22)(q22;q12) | EWSR1-ERG | Ets-like | 10 % | |
t(7;22)(p22;q12) | EWSR1-ETV1 | Ets-like | Rare | |
t(17;22)(q12;q12) | EWSR1-ETV4 | Ets-like | Rare | |
t(2;22)(q33;q12) | EWSR1-FEV | Ets-like | Rare | |
Desmoplastic small round-cell tumor | t(11;22)(p13;q12) | EWSR1-WT1 | Zinc finger | 95 % |
Inflammatory myofibroblastic tumor | t(1;2)(q25;p23) | ALK-TPM3 | Tyrosine kinase | N/A |
t(2;19)(p23;p13) | ALK-TPM4 | Tyrosine kinase | N/A | |
t(2;17)(p23;q23) | ALK-CLTC | Tyrosine kinase | N/A | |
Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 | bZIP | 95 % |
t(12;22)(q13;q12) | EWSR1-ATF1 | bZIP | 5 % | |
Myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1-NR4A3
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|