This article focuses on the cellular, biochemical, and molecular pharmacology of antifolates and how a basic understanding of the mechanism of action of methotrexate, its cytotoxic determinants, mechanisms of resistance, and transport into and out of cells has led to the development of a new generation of antifolates, a process that continues in the laboratory and in the clinics. New approaches to folate-based cancer chemotherapy are described based on the targeted delivery of drugs to malignant cells.
- •
Antifolates disrupt cellular proliferation by blocking folate-dependent one-carbon biosynthetic and methylation reactions.
- •
Antifolates form active polyglutamate derivatives that are retained in tumor cells and result in sustained inhibition of their target enzymes.
- •
Antifolates currently approved for cancer treatment include methotrexate and pralatrexate, inhibitors of dihydrofolate reductase, and pemetrexed, which, in its polyglutamate forms, targets thymidylate synthase and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase.
- •
Novel folate analogues and conjugates, currently in clinical trial, use membrane folate receptors for transport into tumor cells by an endocytic mechanism.
The antifolates were the first class of antimetabolites to enter the clinics 65 years ago. These agents disrupt the metabolic pathways that require one-carbon moieties supplied by the B9 folate vitamins that they resemble. Although renewing tissues of the bone marrow and intestinal tract are also folate-dependent and are sites of antifolate toxicity, the clinical utility of antifolates was established with the identification of doses and schedules of administration that provided sufficient selectivity to make these drugs effective in the treatment of cancer and inflammatory disorders. Many of the early key preclinical studies that defined the pharmacologic properties of this class of drugs, and treatment strategies, were conducted in vitro and in vivo in mice using murine leukemia cell lines.
The first antifolate in the clinic was aminopterin. Its introduction, as first reported in the New England Journal of Medicine in June 1948, was greeted with great enthusiasm when this agent was shown to produce, for the first time, remissions in children with acute lymphoblastic leukemia. Although these remissions were short lived, the activity of this agent established that this disease was treatable and provided optimism that this and other malignant diseases would be conquerable with cancer chemotherapeutics in the future. For reasons not fully understood, but attributed to the unpredictable toxicity of aminopterin, this drug was replaced with methotrexate (MTX) in the early 1950s, an antifolate less potent than aminopterin but with what was considered to be a more favorable therapeutic index.
Despite its early clinical success, an understanding of the mechanism of action of MTX evolved slowly over the ensuing decades. Likewise, the efficacy and selectivity of leucovorin rescue that allowed the safe administration of high doses of MTX was established entirely empirically and, even today, the basis for the selectivity of this regimen is not widely appreciated nor fully understood. The lack of a basic understanding of the biochemical and molecular pharmacology of MTX hampered efforts to develop subsequent generations of antifolates that would lead to the realization of the full clinical potential of this class of drugs. Hence, it was more than 50 years after the introduction of MTX that the second antifolate, pemetrexed, was approved in 2004 for the treatment of mesothelioma and subsequently non–small cell lung cancer. This was followed by the approval of pralatrexate in 2009 for the treatment of cutaneous T-cell lymphoma.
This article focuses on the cellular, biochemical, and molecular pharmacology of antifolates and how a basic understanding of the mechanism of action of MTX, its cytotoxic determinants, mechanisms of resistance, and transport into and out of cells has led to the development of a new generation of antifolates, a process that continues both in the laboratory and in the clinics. Finally, new approaches to folate-based cancer chemotherapy are described based on the targeted delivery of drugs to malignant cells. These approaches include (1) the development of folate analogues transported by routes selectively expressed or active in tumor cells and (2) the selective endocytosis of folic acid, linked to cytotoxics, into tumor cells that express folate receptors. The evolution of antifolates, folate receptor targeted drugs, and membrane transport of folates and antifolates have been the subject of recent reviews.
Antifolates achieve their pharmacologic effects by their perturbations of folate metabolism
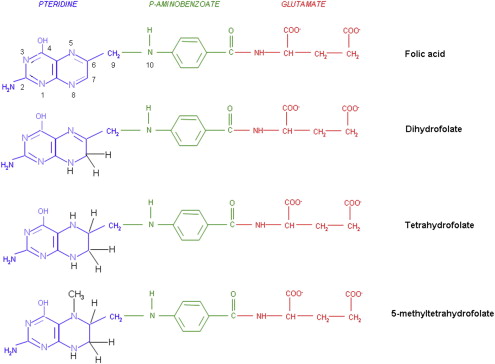
The structures of the B9 vitamins in their oxidized and reduced forms are illustrated in Fig. 1 . Folic acid is not a physiologic folate but is an important source of folates ingested because it is added as a supplement to foods and it is the predominant form of folate in vitamin supplements. The major dietary folate in nature, 5-methyltetrahydrofolate (5-methylTHF), is absorbed in the proximal small intestine by a highly specific transport mechanism, the proton-coupled folate transporter (PCFT, see later discussion), following which it is delivered to the liver via the hepatic portal vein. There, large amounts of 5-methylTHF accumulate as polyglutamate derivatives (see later discussion). Following conversion back to the monoglutamate form, this folate exits hepatic cells to circulate in the blood for delivery via the reduced folate carrier (RFC, see later discussion) to peripheral tissues. There it provides its methyl group to homocysteine for the synthesis of methionine with the generation of a tetrahydrofolate moiety in a vitamin B12 requiring reaction mediated by the enzyme methionine synthase ( Fig. 2 ). The tetrahydrofolate generated then proceeds, through a variety of reactions, to acquire another carbon at various oxidation states at the N 5 , N 10 , or shared between both positions, to form a family of tetrahydrofolate cofactors. These one-carbon derivatives sustain key biosynthetic reactions within cells, such as the addition of a single carbon in the synthesis of thymidylate and 2 carbons in the synthesis of purines, which are both required for the synthesis of DNA and RNA. Methionine goes on to form S-adenosyl methionine, which then mediates a variety of methylation reactions, including the methylation of cytosines within DNA, which is a key factor in the regulation of transcription and activities of oncogenes and tumor suppressor genes. In the absence of vitamin B12, the use of 5-methylTHF cannot occur; 5-methylTHF is trapped so that neither its one-carbon nor tetrahydrofolate moieties are available for biosynthetic reactions. Proliferating tissues, with a high requirement for folates, are the most susceptible to folate deficiency and to drugs that block these folate-dependent pathways.
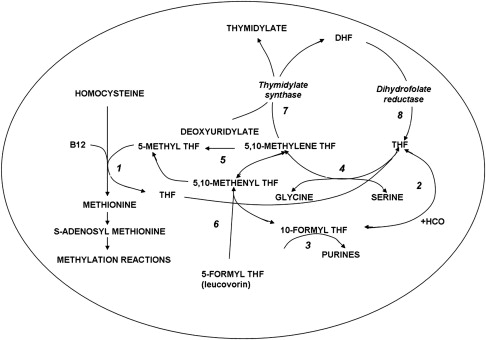
In the synthesis of methionine and purines, the tetrahydrofolate molecule remains intact and another carbon moiety can be acquired to be used again in one-carbon reactions (see Fig. 2 ). However, in the synthesis of thymidylate from deoxyuridylate that requires 5,10-methylene tetrahydrofolate (5,10-methyleneTHF), tetrahydrofolate is oxidized to dihydrofolate. In proliferating cells, this reaction is so rapid that tetrahydrofolate cofactors would rapidly interconvert to 5,10-methyleneTHF, followed by oxidization to dihydrofolate, in minutes if it were not for the enzyme, dihydrofolate reductase (DHFR), which rapidly reduces dihydrofolate back to tetrahydrofolate to sustain the pool of tetrahydrofolate cofactors.
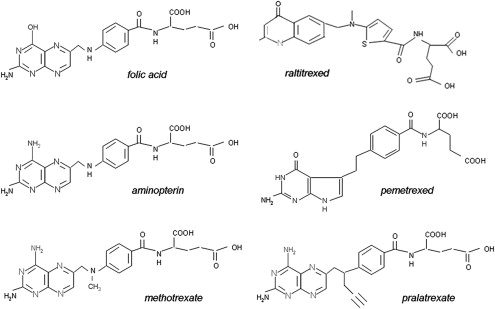
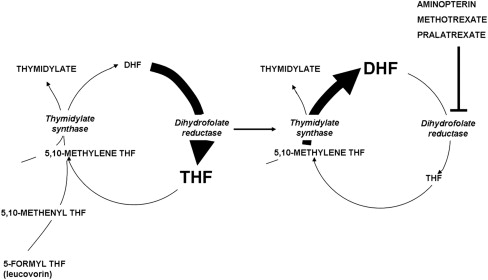
Fig. 3 illustrates the structures of aminopterin and MTX. The former differs from folic acid only in the substitution of an amino for a hydroxyl group at the N 4 position of the pteridine ring. MTX differs from aminopterin in having the addition of a methyl group at the N 10 position in a bridge between the pteridine and p-amino-benzoic acid moieties. These structural differences from folic acid confer on these analogues an extremely high affinity for DHFR. The primary action of aminopterin and MTX, the classical antifolates, is the inhibition of this enzyme. The 4-amino antifolates are among the most potent enzyme inhibitors known. When the interaction between the drug and its target enzyme is evaluated in cell-free systems, the inhibition constant is approximately 5 pM. This value is comparable to what can be achieved with inhibitors designed to bind within the transition state of the catalytic sites of enzymes. However, micromolar levels of MTX are required to inhibit this enzyme within cells. There are 2 reasons for this discrepancy: (1) DHFR is present in cells in great excess so that inhibition of more than 95% of the enzyme is required to begin to suppress tetrahydrofolate synthesis and (2) as illustrated in Fig. 4 , the substrate for this reaction, dihydrofolate, builds to high levels behind the block as tetrahydrofolate cofactors interconvert and cycle to 5,10-methyleneTHF, which is oxidized via thymidylate synthase. Dihydrofolate levels become so high that they compete with MTX for the small percentage of enzyme that is sufficient to support tetrahydrofolate synthesis. To overcome this inhibition and achieve saturation of the enzyme requires relatively high, micromolar, intracellular levels of MTX. Indeed, if after sufficient levels of MTX are present within the cell to completely block this reaction, cells are placed in an MTX-free environment, the drug rapidly leaves the cell, a small component of MTX bound to DHFR is displaced by the high levels of dihydrofolate, tetrahydrofolate synthesis resumes, tetrahydrofolate cofactors rapidly increase to normal levels, and dihydrofolate decreases to its usual very low levels leaving greater than 95% of the enzyme still associated with MTX. This sequence of events occurs within cancer cells as MTX blood levels increase and decrease after the administration of the drug. Hence, a potent inhibitor of DHFR in a cell-free system is a competitive and rapidly reversible inhibitor when interacting with its target enzyme within the dynamic environment of the cell where pools of substrate and products can undergo large changes when enzyme activities are perturbed.
Based on this understanding of the critical elements of MTX interactions within cells, it becomes clear that a major determinant of the activity of this agent is the intracellular level achieved. There are 2 determinants of the cell antifolate level: (1) membrane transport and (2) metabolic conversion to derivatives that remain potent inhibitors of DHFR but are retained within the cell.
Pharmacologic consequences of the formation of methotrexate polyglutamate derivatives within cells
Physiologic folates form polyglutamate derivatives that are retained and build to high levels within cells; these are usually preferred substrates for tetrahydrofolate cofactor-requiring enzymes. In a series of reactions, mediated by folylpolyglutamate synthetase (FPGS), glutamate molecules are added successively at the γ-carboxyl moiety to form a peptide chain of up to 6 to 8 glutamate residues. Antifolates undergo the same reaction and this has profound pharmacologic ramifications. The polyglutamation of antifolates (as seen for MTX in Fig. 5 ) converts these drugs from a form (monoglutamate) that is a good substrate for a variety of folate export processes, to forms that are not, leading to the retention and buildup of high levels of polyglutamate derivatives in tumor cells. In the case of MTX, these derivatives are at least as potent inhibitors of DHFR as the monoglutamate. The retention of MTX polyglutamate derivatives within cells, after antifolate blood levels decrease, results in sustained inhibition of this enzyme for long intervals. In the case of pemetrexed, polyglutamation results in derivatives with a much higher affinity for its target enzymes than the monoglutamate and this, along with prolonged retention in tumor cells, allows regimens in which this agent can be administered every 3 weeks (see later discussion).
A key factor in the efficacy of any antineoplastic agent is its selectivity: the extent to which it kills malignant cells while sparing susceptible host tissues. In the case of MTX, the major toxicities are caused by its effects on intestinal and bone marrow cells. The basis for this selectivity seems to be due, at least in part, to the lesser accumulation of its polyglutamate derivatives in these normal replicating tissues in comparison with susceptible tumor cells. Hence, when the drug is administered intravenously in a pulse, MTX is transported into tumor cells where polyglutamate derivatives are synthesized and accumulate. When the concentration of MTX in the blood decreases below a critical level, formation of polyglutamates cease but the polyglutamate derivatives already synthesized are retained and produce prolonged suppression of DHFR. On the other hand, while MTX monoglutamate builds within intestinal and bone marrow cells when the concentration of MTX in the blood is high, as the MTX blood level decreases, MTX monoglutamate exits the cells and, in the absence of polyglutamate derivatives, the small component of DHFR necessary to meet cellular demands for tetrahydrofolate is rapidly activated. For a detailed analysis of the perturbations of cellular folate pools in response to 4-amino antifolates and the impact of the formation of polyglutamate derivatives, the reader is referred to an earlier review.
Pharmacologic consequences of the formation of methotrexate polyglutamate derivatives within cells
Physiologic folates form polyglutamate derivatives that are retained and build to high levels within cells; these are usually preferred substrates for tetrahydrofolate cofactor-requiring enzymes. In a series of reactions, mediated by folylpolyglutamate synthetase (FPGS), glutamate molecules are added successively at the γ-carboxyl moiety to form a peptide chain of up to 6 to 8 glutamate residues. Antifolates undergo the same reaction and this has profound pharmacologic ramifications. The polyglutamation of antifolates (as seen for MTX in Fig. 5 ) converts these drugs from a form (monoglutamate) that is a good substrate for a variety of folate export processes, to forms that are not, leading to the retention and buildup of high levels of polyglutamate derivatives in tumor cells. In the case of MTX, these derivatives are at least as potent inhibitors of DHFR as the monoglutamate. The retention of MTX polyglutamate derivatives within cells, after antifolate blood levels decrease, results in sustained inhibition of this enzyme for long intervals. In the case of pemetrexed, polyglutamation results in derivatives with a much higher affinity for its target enzymes than the monoglutamate and this, along with prolonged retention in tumor cells, allows regimens in which this agent can be administered every 3 weeks (see later discussion).
A key factor in the efficacy of any antineoplastic agent is its selectivity: the extent to which it kills malignant cells while sparing susceptible host tissues. In the case of MTX, the major toxicities are caused by its effects on intestinal and bone marrow cells. The basis for this selectivity seems to be due, at least in part, to the lesser accumulation of its polyglutamate derivatives in these normal replicating tissues in comparison with susceptible tumor cells. Hence, when the drug is administered intravenously in a pulse, MTX is transported into tumor cells where polyglutamate derivatives are synthesized and accumulate. When the concentration of MTX in the blood decreases below a critical level, formation of polyglutamates cease but the polyglutamate derivatives already synthesized are retained and produce prolonged suppression of DHFR. On the other hand, while MTX monoglutamate builds within intestinal and bone marrow cells when the concentration of MTX in the blood is high, as the MTX blood level decreases, MTX monoglutamate exits the cells and, in the absence of polyglutamate derivatives, the small component of DHFR necessary to meet cellular demands for tetrahydrofolate is rapidly activated. For a detailed analysis of the perturbations of cellular folate pools in response to 4-amino antifolates and the impact of the formation of polyglutamate derivatives, the reader is referred to an earlier review.
The membrane transport of antifolates mediated by facilitative carriers and export pumps
The physiologic folates and most antifolates are bivalent anions that diffuse poorly across cell membranes, and only low levels of folates are present in the diet and in the blood. Hence, specific processes are required to achieve their efficient transport across the epithelia and into systemic cells ( Fig. 6 ). Transport across the apical-brush border membrane of the duodenum and proximal jejunum is mediated by the PCFT, a process that functions optimally at the low pH found at the microenvironment of the surface of the villi of these tissues. Its critical role in this process was established with the demonstration that there are loss of function mutations in this gene in the autosomal hereditary disorder, hereditary folate malabsorption. PCFT is expressed in many normal tissues but has limited function because of the ambient neutral pH. Besides the proximal small intestine, PCFT is highly expressed in the liver, choroid plexus, brush-border membrane of the proximal renal tubule, and in a broad spectrum of solid tumors. Transport into systemic tissues is mediated by the RFC, a process that functions optimally at the neutral pH sustained in most tissues. RFC is expressed in all normal cells and is the major route of 5-methylTHF transport into these tissues. RFC is also a major route of transport of MTX and most other antifolates into tumor cells.
MTX, raltitrexed, and pemetrexed share comparable affinities for RFC, with influx K m s of approximately 5 to 7 μM at the optimal pH of this transporter, 7.4. The affinity of RFC for the natural isomer of 5-methylTHF is greater. The affinity of RFC for pralatrexate is 3 to 4 times greater than for MTX. The affinities and maximum transport velocities mediated by PCFT are highest at a pH of approximately 5.5 and decrease as the pH is increased. At its optimal low pH, the affinity of PCFT is greatest for pemetrexed (K m ∼0.2–0.5 μM) but still quite good for MTX (K m ∼1.5 μM). As the pH is increased, transport is much better preserved for pemetrexed than MTX or raltitrexed.
Both RFC and PCFT are facilitative carriers that mediate folate and antifolate entry (influx) and exit (efflux) from cells. The extent to which these processes are unequal will determine the final concentration gradient achieved across the cell membrane. PCFT contains 2 substrates binding sites: one for a proton the other for a folate/antifolate. This symporter uses the transmembrane proton gradient to achieve concentrative folate transport into cells. RFC contains only one substrate-binding site, which is for a folate/antifolate. However, a variety of structurally unrelated organic anions can compete for this site and can use this carrier, the most potent of which are the organic phosphates. This transporter uses the high concentration of organic phosphates within cells to achieve concentrative folate/antifolate transport into cells. Hence, although both transporters are bidirectional and are capable of transporting folates/antifolates into and out of cells, export mediated by PCFT is suppressed by the neutral pH within the intracellular compartment. Likewise, the high levels of organic phosphates within cells suppress the export mediated by RFC, an antiporter. These asymmetrical effects by protons and organic phosphates, respectively, result in concentrative (uphill) folate/antifolate transport into cells.
Antifolates are also substrates for members of the family of multidrug resistance–associated proteins (MRPs) and the breast cancer–resistance protein (BCRP). These ATP-dependent exporters oppose the concentrative effects of RFC or PCFT to suppress the level of monoglutamate antifolates that accumulate within cells and thereby slow the formation of polyglutamate derivatives. When these exporters are inhibited, intracellular MTX levels increase. Once polyglutamate derivatives form, they are no longer substrates for most of these exporters. However, the lower polyglutamate derivatives of MTX are modest substrates for some MRPs and BCRP. The level of monoglutamyl antifolates that accumulate within cells is determined by the net effect of the exporters and the carrier mediated concentrative transporters. Cumulatively, these factors influence the rate and extent of accumulation of antifolate polyglutamate derivatives within cells. Although overexpression of MRPs and BCRP can induce resistance to antifolates, it is unclear whether this contributes to resistance to these agents in the clinics.
Membrane transport of MTX mediated by RFC is an important determinant of its cytotoxicity. The higher the affinity of RFC for this drug, the higher the free levels achieved within the intracellular water and the greater the rate and extent of formation of its polyglutamate derivatives. Impaired membrane transport mediated by RFC is an established mechanism of intrinsic and acquired resistance to MTX (see later discussion). In the absence of RFC function, the only way antifolate activity can be sustained is if the drug can enter tumor cells by passive diffusion or by another transport route. However, to achieve the former for a hydrophilic drug or the latter via PCFT, which operates inefficiently at neutral pH, requires high drug levels with a high potential for toxicity to normal tissues. The membrane transport of folates and antifolates has been the subject of recent reviews.
High-dose MTX with leucovorin rescue
With the recognition that MTX resistance in experimental tumor systems was often associated with impaired RFC-mediated transport across the cell membrane and with the assumption that delivery to cells within solid tumors is limited by a compromised vasculature, an approach was developed to circumvent these limitations that would permit the safe administration of high doses of MTX.
The rationale for high-dose MTX is that high blood and extracellular drug levels facilitate diffusion of drug into the core of solid tumors and passive diffusion across the tumor cell membrane, where carrier-mediated transport might be compromised. Protection of normal tissues is achieved by the subsequent administration of low doses of 5-formyltetrahydrofolate (leucovorin) following treatment with MTX. Presumably, leucovorin has access to bone marrow and intestinal cells via an intact vascular supply to these normal tissues, and intact membrane transport, whereas delivery to tumor cells is limited by the low blood levels of the rescue agent, the compromised vascular supply, and impaired transport across the tumor cell membrane. Additionally, as the MTX blood level decreases, leucovorin concentrations in the blood are sufficiently high to competitively inhibit MTX transport into normal cells via RFC (a transport mechanism that MTX and leucovorin share), so that the MRP exporters pump the drug, unopposed, out of these cells. Although high-dose MTX regimens were implemented in the 1960s, these regimens remain part of the treatment arsenal for several malignancies (see later discussion).
A further understanding of the selectivity of high-dose MTX with leucovorin rescue emerged several decades after the introduction of this regimen and relates to the differential formation of MTX polyglutamate derivatives in tumors versus normal bone marrow and intestinal precursors. This understanding was based on the revelation that polyglutamate derivatives of MTX are not only potent inhibitors of DHFR but also direct inhibitors of tetrahydrofolate-requiring enzymes: thymidylate synthase and one of the enzymes required for the synthesis of purines, 5-aminoimidazole-4-carboxamide ribotide transformylase (AICAR transformylase). These direct inhibitory effects block the use of the one-carbon moiety of leucovorin for thymidylate and purine synthesis in tumors; however, use is unimpeded in normal tissues that lack comparable levels of these derivatives. An additional factor is the interconversion of leucovorin to other tetrahydrofolate cofactors and ultimately to dihydrofolate polyglutamates that displace MTX from, and thereby activate, DHFR in cells that do not contain MTX polyglutamate derivatives.
There are a variety of ways of administering high doses of MTX, including pulses of the drug, or continuous infusions for up to 42 hours, following which low-dose leucovorin is begun. Specific regimens of administration have been developed and parameters have been designated to assure the safety of these protocols based on the expected renal excretion of MTX and the decline in the MTX blood level. Any compromise of renal function and glomerular filtration, which can occur after high-dose MTX, delays the clearance of the drug and requires prolongation of leucovorin rescue until the MTX concentration in the blood decreases to a safe level.
Related posts:
 Topoisomerase 1 Inhibitors and Cancer Therapy
Topoisomerase 1 Inhibitors and Cancer Therapy
 Targeting Angiogenesis in Gynecologic Cancers
Targeting Angiogenesis in Gynecologic Cancers
 Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
 mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
Antimitotic Inhibitors
mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
Antimitotic Inhibitors
 Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


