Fig. 6.1
The androgen receptor pathway and novel targeted agents
The activated AR can also function as a transcription repressor through its antagonistic interaction with transcription factors (e.g., specificity protein 1 [SP1] and Sma and Mad related protein-3 [SMAD3]) and its direct recruitment of transcriptional repressors (e.g., Alien and small heterodimer partner [SHP]) [10]. This dual functionality has important implications in terms of the receptor’s response to androgen deprivation therapy (ADT), currently the standard treatment for metastatic CRPC. Androgen depletion initially leads to an interruption of the AR signaling pathway, but the prostate cancer cell has developed multiple mechanisms to adapt to this deprivation and regain its metabolic function. Thus, a better understanding of the basic mechanisms and function of the AR-ligand pathway, as well as its ability to adapt to a clinical castrate state, has led to recent advances in drug discovery and resulted in improved outcomes in this patient population.
AR Signaling in CRPC
As patients undergo castrating therapy, their tumors undergo adaptive changes by which the AR molecule retains its role as the primary engine of tumor growth despite treatment. AR overexpression [11], mutations within the AR, intracrine synthesis of androgen [12, 13], AR splice variants [14, 15], and activation of alternative pathways such as the phosphatidylinositol-3-OH kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway [16] are some of the means by which the AR continues to signal in the castrate state.
AR gene amplification and protein overexpression are thought to be two of the main mechanisms driving CRPC. In xenograft models, AR overexpression was found to be both necessary and sufficient to induce CRPC [17]. In addition, studies utilizing expression profiling analysis and fluorescence in situ hybridization have identified AR amplification and protein overexpression in approximately 30 % of recurrent prostate cancer specimens [18–20]. A number of androgen-related genes have been found to be upregulated after ADT; this process is thought to lead to sensitization of the tumor cell to lower levels of androgens and reactivation of the AR-ligand pathway thus allowing CRPC to proliferate in an androgen depleted environment [11, 13, 19, 21–25]. Furthermore, overexpression of the AR has also been found to be a potential driver of first-generation antiandrogen agonist activity [17].
Mutations within the LBD and NTD of the AR have also been identified as mechanisms of resistance in CRPC. Specifically, mutations within the LBD can confer resistance to antiandrogen therapy and allow for decreased specificity of AR–ligand interaction, thus enabling alternative steroidal molecules (e.g., estrogens, corticosteroids, and progesterone) to activate the AR pathway [26, 27]. Additionally, molecular dissection of the NTD transactivation unit 5 domain has demonstrated that a WxxLF motif was fully responsible for ligand-independent activity [28].
The recognition of AR splice variants in tissue from patients with CRPC has provided more insight into the resistance mechanisms driving this disease [29, 30]. Although AR splice variants lack an LBD, they contain an intact NTD and DBD and have been shown to be active in the absence of androgens [31, 32]. Regulation of AR splice variants remains poorly understood; however, increased expression has been seen in prostate cancer cell lines that have been introduced to an androgen antagonist [33]. At least seven splice variants of the AR have been uncovered to date, and two of these variants have up to a 20-fold higher expression in CRPC compared to castration-sensitive prostate cancer; and higher expression of one variant, AR-V7, was seen to predict biochemical recurrence following definitive local therapy in castration-sensitive disease [34].
Finally, kinase pathway activation is yet another mechanism central to the development of CRPC. The PI3K/AKT/mTOR pathway plays an important role in tumorigenesis and therapy resistance in multiple malignancies, including prostate cancer, and activation of this pathway is strongly associated with prostate cancer progression [35–37]. PI3K/AKT/mTOR pathway alterations are seen in up to 43 % of primary prostate cancer cases and up to 100 % in metastatic disease, with loss of PTEN (phosphatase and tensin homolog), a tumor suppressor gene, accounting for roughly 40 % of cases [35, 37]. Loss of PTEN has been correlated with resistance to castration in in vitro models, though this effect is not absolute [38, 39].
All of these mechanisms—gene amplification, protein overexpression, constitutive activation, and promiscuous mutation—are mechanisms of resistance in CRPC. All of these, as well, point to the AR as a prime target for treating CRPC to yield true clinical benefits.
First-Generation Antiandrogens
First-generation nonsteroidal antiandrogens include flutamide, nilutamide, and bicalutamide. These nonsteroidal agents compete with endogenous androgens for binding within the LBD, resulting in a conformational change in the AR that inhibits transcriptional activity [40].
Clinically, these drugs have traditionally been used to prevent flare, which is seen in the setting of initial gonadotropin-releasing hormone (GnRH) use as well as in combined androgen blockade (CAB) therapy. Flare occurs in roughly 10 % of patients starting GnRH agonist therapy and is related to the surge of androgens during the first week of treatment; this surge is potentially responsible for a rise in PSA and worsening of clinical symptoms [41]. The flare phenomena in patients with advanced disease can lead to worsening back pain, urinary obstruction, and other negative symptoms, and is routinely suppressed with the use of concurrent administration of a GnRH analog and an antiandrogen agent. These drugs have also been used chronically in conjunction with surgical or medical castration as part of CAB therapy, a controversial strategy whose benefits remain ambiguous. At least three large meta-analyses evaluating the survival advantage of CAB have been published. One analysis showed a 10 % improvement in overall survival after CAB (relative risk = 0.90; 95 % confidence interval [CI] 0.79–1.00), another revealed no survival advantage, and the third found no benefit at 2 years (20 trials, hazard ratio [HR] = 0.970; 95 % CI 0.866–1.087) but a statistically significant difference in survival at 5 years (10 trials, HR = 0.871; 95 % CI 0.805–0.942) [42–44]. Therefore, CAB therapy might potentially benefit a subset of patients, although the relative effect on all patients with prostate cancer remains uncertain.
Whether antiandrogens are used for flare prevention or as a component of CAB, most patients who initially respond to ADT eventually progress to CRPC via the mechanisms described above. Specific mutations within the AR have also explained tumor growth in the setting of antiandrogen use [45, 46]. For example, a novel mutation within codon 741 of the AR LBD has been shown to convert bicalutamide from an antagonist to an agonist, while flutamide acquires agonistic properties when exposed to mutations on codons 874 and 877 [47]. As a result, a subclass of patients with these mutations will develop a paradoxical decline in their PSA level and tumor size when conventional antiandrogens are withdrawn, indicating an agonistic property [48–50]. Specifically, a Southwest Oncology Group clinical trial (SWOG 9426) examined the antiandrogen withdrawal phenomenon using a variety of first-generation antiandrogens and found that 21 % of the 210 patients enrolled had a confirmed PSA decline of ≥50 % at the termination of antiandrogen use. Of these patients, 64 % were on flutamide, 32 % on bicalutamide, and 3 % on nilutamide [48]. A phase 3 clinical trial, CALGB 9583, compared patients undergoing antiandrogen withdrawal either alone or in combination with ketoconazole. That study found that 15 of 132 patients (11 %) of patients who underwent antiandrogen withdrawal alone demonstrated a PSA decline of ≥50 %, with a median time to PSA progression of 5.9 months [51]. The antiandrogen withdrawal phenomenon highlights the inability of first-generation antiandrogens to durably repress AR signaling, as they can serve as partial agonists despite initial repression of signaling. This key weakness spurred a search for new drugs that did not have partial agonistic activity and thus repressed the AR more fully and more durably.
BMS-641988
BMS-641988 is a competitive inhibitor of the AR found to partially overcome some of the pitfalls of first-generation antiandrogen agents. In preclinical studies using the CWR-22-BMSLD1 human prostate cancer xenograft model, BMS-641988 displayed an increased efficacy over bicalutamide, with an average tumor growth inhibition of >90 % versus <50 %, respectively. In addition, researchers administered BMS-641988 to a bicalutamide-refractory CWR-22-BMSLD1 model and found that BMS-641988 significantly delayed tumor growth, whereas the group that continued bicalutamide therapy had tumors that grew progressively. However, once BMS-641988 therapy was stopped, tumors in that group restored their capacity to grow reflecting a more cytostatic rather than cytocidal effect [52]. Its ability to bind to the wild-type AR with a 20-fold higher affinity than bicalutamide and display activity towards the mutant AR eventually led to BMS-641988 to be studied in a clinical trial setting.
BMS-641988 was investigated in a phase I dose-escalation study involving 61 men with metastatic CRPC (Table 6.1). A majority of patients enrolled on the trial (65 %) had undergone three or more treatment regimens, including 16 (26 %) who had received prior chemotherapy with docetaxel. A dose range between 5 and 140 mg per day was evaluated, with no patients remaining on therapy at the completion of the trial. The two most common adverse events were fatigue (25 %) and gastrointestinal events (31 %), all of which were grade 1 or 2. However, one patient had a grade 3 seizure event while receiving 60 mg of BMS-641988, which resulted in termination of the trial. At the completion of the trial, 10 of the 61 patients (16 %) had a >30 % decline in PSA. Of 23 patients with measurable disease, one (4 %) had a partial response and 17 (74 %) had stable disease on imaging. In addition, after termination of treatment with BMS-641988, some patients had a decline in their PSA, revealing a possible antiandrogen withdrawal response and implying that BMS-641988 might have both agonistic and antagonistic properties. Therefore, as a result of its serious adverse event profile and failure to meet basic efficacy criteria, the trial was terminated and the development of BMS-641988 was discontinued [53].
Table 6.1
Clinical trials of androgen receptor targeted agents for CRPC
Class | Agent | Phase | Treatment arms | Primary end point | Remarks and patient population | Reference/ClinicalTrials.gov identifier |
|---|---|---|---|---|---|---|
Targeted agents | Enzalutamide (MDV3100) | III | Double-blinded placebo controlled: enzalutamide vs. placebo | OS | AFFIRM trial, post-docetaxel-based chemo mCRPC. Trial stopped early due to benefit | [65], NCT00974311 |
Enzalutamide (MDV3100) | III | Double-blinded placebo controlled: enzalutamide vs. placebo | OS and rPFS | PREVAIL trial, pre-chemo mCRPC. Trial stopped early due to benefit | [66], NCT01212991 | |
BMS-641988 | I | Randomized dose-escalation | Safety and tolerability | Study closed due to limited antitumor affect and seizure activity | [53] | |
ARN-509 | I/II | Non-randomized, open label | PSA response | Three arm: treatment-naïve nonmetastatic CRPC vs. treatment naïve mCRPC vs. post-abiraterone mCRPC | [77], NCT01171898 | |
ARN-509 | Ib | Open label: ARN-509 + abiraterone + prednisone | MTD/RP2D when administered with abiraterone | mCRPC | NCT01792687 | |
ARN-509 | II | Randomized, open label, three arm: ARN-509 alone vs. ARN-509 + LHRH antagonist vs. LHRH antagonist alone | Mean change in QoL measured by total FACT-P score | Biochemical-relapsed hormone-sensitive prostate cancer | NCT01790126 | |
ARN-509 | III | Randomized, placebo controlled | Metastasis-free survival | SPARTAN trial, nonmetastatic CRPC | NCT01946204 | |
ODM-201 | I/II | Randomized, open label | Safety and tolerability | ARADES trial, dose-escalation study with a randomized phase II expansion component, mCRPC | ||
AZD3514 | I | Randomized, open label | Safety and tolerability | mCRPC | NCT01162395 | |
Hsp90 | 17-AAG | II | Open label | PSA response | Grade 3 toxicity included fatigue, lymphopenia, and back pain | [88] |
Ganetespib (STA-9090) | II | Single arm | PFS | Post-chemo mCRPC | NCT01270880 | |
AT13387 | I/II | Randomized, open label, two arm: AT133887 + abiraterone vs. AT13387 alone | Phase A: safety, tolerability, optimal dosing; Phase B: response rate per PCWG2 [59] and CTC count between single agent and combo | Post-abiraterone mCRPC | NCT01685268 | |
Hsp27 | OGX-427 | II | Randomized, open label, two arm: OGX-427 + prednisone vs. prednisone alone | PFS (per PCWG2) | Chemo-naive mCRPC | NCT01120470 |
Antisense oligonucleotides | Custirsen (OGX-011) | III | Randomized, open label, two arm: docetaxel + prednisone + OGX-011 vs. docetaxel + prednisone | OS | SYNERGY trial, chemonaïve mCRPC Study completed. Did not meet primary endpoint of OS | NCT01188187 [112] |
Custirsen (OGX-011) | III | Randomized, open label, two arm: cabazitaxel + prednisone + OGX-011 vs cabazitaxel + prednisone | OS | AFFINITY trial, post 1st line chemo, mCRPC | NCT01578655 | |
EZN-4176 | I | Non-randomized, open label | MTD and safety profile | AR mRNA antagonist, mCRPC | [111] | |
Epigenetic therapies | Azacitidine | I/II | Non-randomized, open label | MTD and safety profile | mCRPC previously treated with docetaxel | NCT00503984 |
Novel targets | TOK-001 | II | Single arm open label | MTD, safety profile, and PSA response | ARMOR2 trial, mCRPC. Three mechanisms of action: inhibits CYP17, antagonizes testosterone binding to AR, degrades AR protein | NCT01709734 |
Enzalutamide
Enzalutamide, formerly known as MDV3100, is a novel oral nonsteroidal AR antagonist developed to target oncogenic alterations associated with resistance to the first-generation antiandrogens. Compared to first-generation agents and BMS-641988, enzalutamide has a greater affinity towards the AR, displays no agonistic properties, and has a distinct ability to inhibit nuclear translocation, DNA binding, and coactivator recruitment (Fig. 6.1). In definitive phase III studies, enzalutamide was found to prolong survival in men with metastatic CRPC, improve their quality of life, and delay time to first skeletal-related event, leading to its approval by the United States Food and Drug Administration in 2012.
Preclinical Development
The development of enzalutamide began with the chemical scaffold molecule RU59063, a nonsteroidal AR agonist, due to its unique selectivity over other nuclear hormone receptors and its high affinity for the AR [54, 55]. Nearly 200 thiohydantoin derivatives of RU59063 were tested for AR agonism and antagonism in human prostate cancer cells engineered to express increased amounts of the AR. After further chemical modifications, including optimization of pharmacokinetic properties such as serum half-life and oral bioavailability, two compounds, diarylthiohydantoins RD162 and MDV3100, were chosen for further investigation [56, 57].
MDV3100 and RD162 both displayed a five- to eight-fold greater affinity for the AR compared with bicalutamide (measured in a competition assay using 16β-[18F]-fluoro-5α-dihydrotestosterone [18F-FDHT]) as well as an increased selectively to the AR (measured in an in vitro fluorescence polarization assay). Additional assays demonstrated that, when bound to the AR, both RD162 and MDV3100 impaired nuclear translocation, DNA binding, and coactivator peptide recruitment, while bicalutamide and DHT did not. Neither RD162 nor MDV3100 activated the wild-type AR or the mutant receptor that converts bicalutamide to a pure agonist (W741C); and in LNCaP/AR xenograft castrate mice, they both induced tumor regression while bicalutamide merely slowed tumor growth [57]. Further, cells treated with either RD162 or MDV3100 showed a five-fold reduction in the ratio of nuclear to cytoplasmic AR compared to bicalutamide-treated cells. Given its favorable drug-like properties and safety profile, MDV3100 was chosen over RD162 for further clinical development.
Phase I/II Trial
Given the encouraging preclinical data, a first-in-man, phase I/II dose-escalation trial was conducted to identify the safety and tolerability profile of MDV3100 as well as to establish the maximum tolerated dose (MTD) [58]. A total of 140 men with metastatic CRPC who were either chemotherapy naïve (n = 65) or had prior taxane exposure (n = 75) were enrolled. Antitumor effects were seen at all doses, including a ≥50 % PSA decline in 78 patients (56 %). Other antitumor effects were evaluated using the Prostate Cancer Clinical Trial Working Group 2 (PCWG2) criteria [59]. Time to biochemical progression, defined as an increase in PSA of ≥25 % from the nadir, was found to be 41 weeks for chemotherapy-naïve patients and 21 weeks for patients who had received prior taxane therapy. There was also a significant difference in radiographic progression between the two groups. For chemotherapy-naïve patients, median time to radiographic progress was not reached, whereas for taxane-pretreated patients it was 29 weeks.
Circulating tumor cells (CTCs) were also collected from 128 patients (91 %) before and after the administration of MDV3100. A cutoff of 5 cells per 7.5 ml of blood was used to determine favorable status (<5 cells) versus unfavorable (≥5 cells) [60]. Of 77 patients who had favorable CTC counts prior to treatment, 70 (91 %) maintained their counts during treatment, whereas 49 % who had unfavorable counts prior to treatment converted to favorable after treatment. CTC status has recently been shown to be an accurate predictor of survival, with a median OS of 21.7 months for patients with favorable pretreatment CTC count versus 11.5 months for patients with unfavorable CTC count [61, 62].
A subset of 22 of the 140 patients participated in an additional experimental biomarker trial, in which positron emission tomography (PET) imaging was used to measure uptake of 18F-FDHT before and after treatment with MDV3100 [58], to assess AR blockade. An androgen analog, 18F-FDHT localizes to tumor tissue and binds to the AR, allowing for direct visualization of the study drug and its target as well as potential identification of treatment-related variations in metastatic sites [63]. Figure 6.2 shows a scan of one patient enrolled on the phase I/II clinical trial evaluating MDV3100. The baseline FDHT-PET scan shows an overexpression of the AR on the left iliac crest through increased uptake of 18F-FDHT. However, after treatment with MDV3100 the 18F-FDHT uptake diminishes, as do the PSA levels (seen on the adjacent graph), revealing that the antiandrogen is inhibiting its target. All 22 patients showed a clear reduction in 18 F-FDHT uptake on post-treatment imaging. Interestingly, uptake of the radiotracer plateaued at a dose of 150 mg of MDV3100, even though higher plasma concentrations of the study drug were found at the higher doses, demonstrating that higher serum concentrations of MDV3100 do not correlate to increased levels of AR binding.
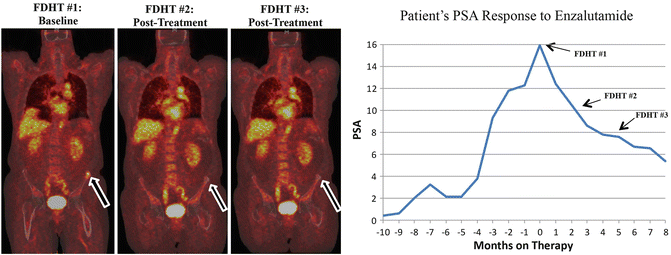
Fig. 6.2
A patient’s response to enzalutamide on FDHT-PET and PSA. The arrows in the figure correlate to a left iliac bone lesion that was noted to have FDHT uptake prior to treatment with enzalutamide. On post-treatment analysis, the FDHT uptake as well as the patient’s PSA declined revealing an on-target and treatment effect with enzalutamide
Toxic effects were graded with the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE version 3.0). Fatigue was the most common grade 3–4 adverse event, seen at doses ≥240 mg and occurring in 11 % of patients; it was also the most common grade 2 adverse event, seen in 27 % of patients. There were also three documented seizures seen at dosages from 360 to 600 mg. Even though antiandrogens as a class have been shown to cause seizures in animal models—through an off-target mechanism inhibiting GABA-A (gamma aminobutyric acid) channels—the patients who experienced seizure activity on trial had underlying comorbidities or were on medications that potentially could have contributed to the event [64]. Based on its tolerability profile, and activity as demonstrated by PSA, conventional imaging, and FDHT responses, a dose of 160 mg per day of MDV3100 was selected to be the optimal dose for further studies [58].
Phase III Trials
As a result of the positive phase I/II clinical trial of MDV3100, a multinational phase III double-blinded placebo-controlled trial (AFFIRM) was initiated to further evaluate the compound, now called enzalutamide, in 1,199 men with chemotherapy-treated CRPC. The trial accrued in 14 months with a 2:1 assignment, and was unblinded by an independent data safety monitoring committee after a planned interim analysis of 520 events was reached. At the time of analysis, a 37 % reduction in the risk of death was seen in the enzalutamide group compared with placebo (HR = 0.63; 95 % CI 0.53–0.75; P < 0.001) [65].
Overall, the AFFIRM trial showed an improvement in OS of 4.8 months (median OS 18.4 months in the enzalutamide group versus 13.6 months in the placebo group). In addition, an advantage of enzalutamide over placebo was seen in all secondary endpoints, including a reduction in PSA level by ≥50 % and improved soft-tissue response rate, time to PSA progression, quality-of-life response rate (using the Functional Assessment of Cancer Therapy-Prostate [FACT-P] questionnaire), radiographic PFS, and time to first skeletal-related event. The most common adverse effects were similar to the phase I/II trial and included fatigue (34 %), diarrhea (21 %), hot flashes (20 %), and seizures (<1 %). As a result, in 2012 enzalutamide was approved for clinical use in the USA in men with metastatic CRPC previously treated with docetaxel (Table 6.1).
More recently, the PREVAIL trial evaluated enzalutamide in 1,717 men with CRPC that progressed despite ADT but who had not yet received chemotherapy. A pre-planned interim analysis was conducted after 540 events (patient deaths) and found benefit in the treatment arm for which the study was terminated. Both primary endpoints, radiographic progression-free survival and overall survival, significantly favored the enzalutamide arm. The rate of radiographic progression-free survival was 65% in the enzalutamide arm versus 14% in the placebo arm (HR=0.19; 95%CI 0.15-0.23) at 12 months of treatment. In addition, 631/872 (72%) patients were alive in the enzalutamide arm compared to 546/845 (63%) patients in the placebo arm for a 29% reduction in the risk of death (HR=0.71; 95% CI 0.60-0.84). Enzalutamide was also found to be superior with respect to all the evaluated secondary endpoints. Fatigue remained the most common adverse effect seen in >33% of patients in the enzalutamide arm which was similar to the AFFIRM trial described above. Other common adverse effects were back pain (27%), constipation (22%) and arthralgia (20%). At the current data-cutoff date of January 15, 2014, 1 patient in both the enzalutamide and placebo arm experienced a seizure [66].
Mechanism of Resistance to Enzalutamide
Enzalutamide has been shown to improve OS, time to first skeletal-related event, and quality of life, but with a median time to radiographic progression of 8 months in the chemotherapy resistant population [65]. Currently, mechanisms of resistance to enzalutamide are under evaluation, although previously described mechanisms in CRPC, including AR overexpression, mutations in the AR, AR splice variants, and bypass pathways, are currently being investigated [10].
One area of potential resistance to enzalutamide is through upregulation of the glucocorticoid receptor, a nuclear receptor in the same family (NR3C) as the AR. In preclinical models it was found to be upregulated in enzalutamide-resistant mice, leading to the expression of a subset of targeted genes that drives prostate cancer growth. Administration of dexamethasone, a glucocorticoid receptor agonist, in an enzalutamide-resistant model suppressed glucocorticoid receptor function while the AR function was partially restored. Furthermore, suppression of the glucocorticoid receptor in mice treated with enzalutamide revealed further suppression in tumor growth compared to mice with normal glucocorticoid receptor function [67].
Another mechanism of resistance to enzalutamide has recently been described through a missense mutation in the LBD of the AR. The AR F876L mutation has been seen in cell lines, xenograft models, and plasma DNA of patients treated with prolonged courses of enzalutamide. The F876L mutation is also associated with an enzalutamide antagonist-to-agonist switch similar to the mutation in codon 741 that is associated with the bicalutamide antagonist-to-agonist switch [47, 68–70]. Mutation F876L has also been shown to reveal resistance to ARN-509 (discussed below) [69, 70].
AR splice variants have also been hypothesized as a possible mechanism of resistance to enzalutamide through their ligand-independent AR transactivation [71]. Enzalutamide is able to antagonize androgen-mediated activation in cell lines that maintain a full-length AR, but the human prostate carcinoma 22Rv1 cell line, which contains an AR splice variant, is able to maintain its growth potential under castrate levels of androgen despite concurrent administration of antiandrogens such as bicalutamide and enzalutamide.
Finally, resistance to enzalutamide may be mediated through loss of PTEN, a tumor suppressor gene, or through activation of the PI3K/AKT/mTOR signaling pathway [72]. The two most frequently activated signaling pathways in prostate cancer are driven by PI3K and AR, and these pathways have been found to regulate each other through reciprocal feedback such that inhibition of either pathway alone causes activation of the other and protects tumor cells from death. The reciprocal feedback between the AR-directed pathway and the PI3K/AKT/mTOR pathway, however, can hypothetically be overcome by a combined treatment approach using an AR antagonist plus a PI3K/AKT/mTOR pathway inhibitor [73, 74]. This approach has been well studied in other malignancies, including breast cancer, with some treatment success [75], and multiple trials are currently studying this combined treatment approach for prostate cancer.
Future Areas of Interest for Enzalutamide
Now that enzalutamide has shown improvement in OS in metastatic CRPC independent of chemotherapy, future areas of research are opening. Enzalutamide is being studied in different prostate cancer populations including castration-sensitive disease. It is also being evaluated in combination with other therapeutic agents, including abiraterone acetate (a CYP17 inhibitor), tivozanib (a VEGF receptor tyrosine kinase inhibitor), and sipuleucel-T (cellular immunotherapy). A phase III randomized clinical trial comparing enzalutamide to enzalutamide plus abiraterone acetate and prednisone in the chemotherapy-naïve population has recently opened as a collaborative study led by the Alliance for Clinical Trials in Oncology (ClinicalTrials.gov Identifier: NCT01949337).
ARN-509
Even as the development of enzalutamide continues to progress into new clinical arenas, newer AR antagonists have shown promising results. ARN-509 is a next-generation antiandrogen with full antagonistic potential, high affinity to the AR (seven- to ten-fold greater affinity compared to bicalutamide), and the ability to limit AR nuclear translocation and AR binding to androgen response elements. When compared to enzalutamide in preclinical models, ARN-509 achieved maximum efficacy at a lower dose (10–30 mg/kg/d for ARN-509 compared to 30–100 mg/kg/d for enzalutamide). In addition, preclinical models showed an approximately two- to four-fold lower steady-state plasma concentration for ARN-509 compared to the equivalent dose of enzalutamide; at the same time, intratumoral levels of ARN-509 and enzalutamide were roughly equivalent, indicating a higher tumor/plasma ratio with ARN-509. Furthermore, after 28 days of therapy with either ARN-509 or enzalutamide, steady-state brain tissue levels were measured in mice and there was a fourfold lower level in the ARN-509 group compared to the enzalutamide group, possibly indicating a lower seizurogenic potential [76].
Related posts:
 Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
Molecular Mechanisms of Prostate Cancer Progression After Castration
Targeting C-Met/VEGF in Castration Resistant Prostate Cancer
 Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
Cytotoxic Chemotherapy (Taxanes and Taxane Combinations)
 Phase I–II Targeted Treatments
Phase I–II Targeted Treatments
 Epigenetics in Castration Resistant Prostate Cancer
Epigenetics in Castration Resistant Prostate Cancer
 Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Radium-223 and Other Radiopharmaceuticals in Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


