Fig. 7.1
Overexpression of TG2 (left panel) in epithelial cells (shown are MCF10A mammary epithelial cells) is associated with their transdifferentiation into mesenchymal looking cells (EMT; a), increased invasion (through the Matrigel; b), ability to form colonies in agarose (anchorage-independent growth; c), cell motility (wound-healing scratch assay; d), and acquisition of stem cell phenotype (enrichment of mammosphere-forming subpopulation; e)
EMT is associated with the acquisition of stem cell (CSCs) properties. The first evidence that EMT and CSC share some common traits was provided by Mani and colleagues (Mani et al. 2008). Subsequent link between EMT and stem cell markers was established in highly aggressive and metastatic basal-like breast tumors (Sarrio et al. 2008). Similarly, the claudin-low subgroup of breast tumors, which display EMT-like characteristics, shares the gene signature of breast CSC (Chang et al. 2005). CSC exhibit intrinsic resistance to chemotherapy; accordingly treatment of MCF-7 breast cancer cells with doxorubicin resulted in selection of cell subpopulation with CSC characteristics (Calcagno et al. 2010). Importantly, these cells showed EMT features and expressed high basal levels of TG2. These observations clearly support a link among TG2, EMT, and CSC. To further test this contention, we directly determined the effect of TG2 expression on CSC functions. Our data demonstrated that TG2 induces CSC phenotype as suggested by increase in CD44+/CD24−/low subpopulation and concomitant decrease in CD326 expression (Kumar et al. 2011). Moreover, TG2 expression enhanced cell motility (Fig. 7.1d) and the ability of mammary epithelial cells to form mammospheres (Fig. 7.1e), self-renewal ability, and plasticity (Kumar et al. 2011). A similar association between TG2, EMT, and CSCs was reported for ovarian cancer cells (Cao et al. 2012). These results reinforce the biological relevance of TG2 in cancer progression not only by facilitating dissemination of tumor cells but also to endow disseminated tumor cells with the ability to generate hierarchically organized tumors at metastatic sites. Indeed, tumor samples from breast cancer patients with a 7-year follow-up revealed that TG2 expression in primary tumors is inversely correlated with recurrence-free and metastasis-free survival (Oh et al. 2011).
7.3 TG2-Regulated Inflammatory Signaling and Cancer Progression
Previous reports have demonstrated that TG2 expression results in constitutive activation of FAK, Akt, and NF-κB (Eckert et al. 2014; Mehta et al. 2010; Agnihotri et al. 2013), the signaling pathways known to play fundamental roles in cancer progression. For example, NF-κB plays a prominent role in cancer progression, stemming from its ability to activate numerous pro-growth, anti-apoptotic, and metastatic genes (DiDonato et al. 2012). Recently, a novel pro-cancer feedback loop where TG2 activates NF-κB and NF-κB, in turn, drives TG2 expression was proposed (Brown 2013) (Fig. 7.2). In cancer cells, this TG2/NF-κB feedback loop may be self-amplifying because high TG2 expression and elevated NF-κB activity are frequently observed in late-stage cancers. As NF-κB activates EMT and controls the expression of numerous pro-survival genes, a phenotypic outcome resulting from the activation of this loop is enhanced cancer progression and metastasis. Most efforts to inhibit NF-κB activation in cancer cells have focused on small molecules that block the IKK kinase activity. In view of the observation that TG2-induced activation of NF-κB is mediated through IKK-independent mechanism (Oh et al. 2011), these inhibitors are unlikely to have any meaningful effect disabling the TG2/NF-κB loop. Moreover, NF-κB-induced transcriptional regulation of HIF-1α is also mediated by the recruitment of TG2/NF-κB complex to the HIF-1α promoter (Kumar and Mehta 2012). Thus, TG2-expressing epithelial cancer cells express high basal level of HIF-1α even under normoxic conditions. Like TG2 and NF-κB, HIF-1α is also considered a negative prognostic factor because of its ability to promote chemoresistance, angiogenesis, invasiveness, metastasis, resistance to cell death, altered metabolism, and genomic stability (DeClerck and Elble 2010). Recently, we showed that TG2-induced HIF-1α expression in mammary epithelial cells results in decreased mitochondrial respiration rates and increased extracellular acidification rates, suggesting a shift in glucose metabolism (Kumar et al. 2014). Indeed, TG2 expression was associated with increased glucose uptake and lactate production and increased expression of glucose metabolic enzymes both at transcript and protein levels (Kumar et al. 2014). Downregulation of constitutive or induced TG2 expression by siRNA could reverse all these processes. Taken together, these results suggest that TG2-induced shift in glucose metabolism represents an important mechanism that protects cancer cells from stressful conditions and promotes their metastatic competence.
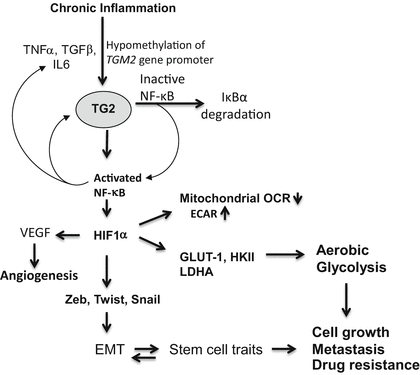
Fig. 7.2
Schematic representation of TG2-regulated inflammatory signaling pathways that contribute to drug resistance and metastatic phenotype in cancer cells. Chronic exposure to inflammatory cytokines (produced by tumor-infiltrating immune cells) results in epigenetic regulation of TG2 and initiates the feedback loop – where TG2 activates NF-κB and NF-κB further increases TG2 expression. HIF-1α is a downstream target of TG2-induced NF-κB. Increased expression of HIF-1α (even under normal oxygen conditions) results in activation of multiple downstream target genes which play critical role in reprogramming shift in glucose metabolism of cancer cells, increased angiogenesis, increased cell survival, and metastasis
Thus, epigenetic regulation of TGM2 in cancer cells can reprogram inflammatory signaling networks (NF-κB activation, expression of HIF-1α, Snail, Twist, and Zeb) that influence many hallmark processes (e.g., by inducing EMT and promoting stem cell traits) to promote drug resistance and metastatic phenotype (Fig. 7.3). These TG2-induced changes endow cancer cells with the ability to disseminate, survive in stressful environments, and regrow at metastatic sites. Indeed, the presence of a canonical CpG island within 5’ flank of TGM2 gene promoter supports epigenetic regulation of TMG2 (Ai et al. 2008). Importantly, doxorubicin-resistant breast cancer cells express high basal level of TG2 due to hypomethylation of TGM2 promoter, whereas doxorubicin-sensitive cells, which show no detectable expression of TG2, have hypermethylated TGM2 promoter (Ai et al. 2008). Accordingly, culture of drug-sensitive cells on 5-azadC restored TG2 expression and reduced their sensitivity to doxorubicin.
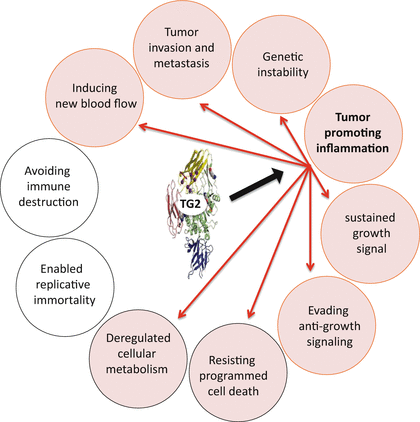
Fig. 7.3
Schematic representation of TG2-regulated inflammatory signaling which endows on cancer cells the ability to metastasize, make new blood vessels, alter glucose metabolism, resist cell death, and evade anti-growth signaling – the important hallmarks of cancer (shown in pink circles)
Taken together, these results suggest that TG2 represents a promising therapeutic target to inhibit multiple pathways that play important roles in promoting drug resistance and progression of metastatic disease. As a proof of concept, published reports have provided compelling evidence that in vivo silencing of TG2 by liposomal-siRNA could effectively inhibit the dissemination of orthotopically growing tumors and render them sensitive to chemotherapeutic drugs in nude mouse mode (Verma et al. 2008; Hwang et al. 2008).
Related posts:
 Prevalence of Gastrointestinal Cancers in India
Prevalence of Gastrointestinal Cancers in India
 Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
 Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
 Apoptosis Pathways in Chronic Lymphocytic Leukemia: Role of the Microenvironment and Therapeutic Strategies
Apoptosis Pathways in Chronic Lymphocytic Leukemia: Role of the Microenvironment and Therapeutic Strategies
 Role of Surfactants in Regulation of Cancer Growth
Role of Surfactants in Regulation of Cancer Growth
 Personalized Therapeutic Strategies for Epithelial Ovarian Cancer
Personalized Therapeutic Strategies for Epithelial Ovarian Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


