This article presents an overview of the PI3K/Akt/mTOR signaling pathway. As a central regulator of cell growth, protein translation, survival, and metabolism, activation of this signaling pathway contributes to the pathogenesis of many tumor types. Biochemical and genetic aberrations of this pathway observed in various cancer types are explored. Last, pathway inhibitors both in development and already approved by the Food and Drug Administration are discussed.
Key points
- •
PI3K/Akt/mTOR pathway:
- ○
Essential role in cell growth, protein translation, survival, and metabolism.
- ○
Activation contributes to pathogenesis of many cancers.
- ○
Second most frequently activated pathway in cancer.
- ○
- •
Preclinical studies and clinical trials ongoing with inhibitors targeting PI3K, PDK-1, Akt, and mTOR.
- •
Rational combination therapies likely key to targeting this pathway effectively.
Cells communicate with each other and respond to environmental conditions through signal transduction pathways. In cancer, deregulation of these pathways results in altered responses, such as increased cell survival and proliferation under conditions that would usually promote cell death or cell cycle arrest. The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian Target of Rapamycin (mTOR) signaling pathway assimilates both intracellular and extracellular signals to control cell metabolism, growth, proliferation, and survival. Activation of PI3K/Akt/mTOR signaling contributes to the pathogenesis of many tumor types, suggesting that targeted inhibition of individual players in this pathway, including PI3K, phosphoinositide-dependent kinase-1 (PDK-1), Akt, and mTOR, is a potential strategy for cancer therapy. This article offers an overview of the pathway, a review of the biochemical and genetic aberrations of the pathway observed in cancer, and a description of pathway inhibitors that are approved and in clinical development.
PI3K/Akt/mTOR signaling pathway
Initiation of signaling through the PI3K/Akt/mTOR pathway occurs through several mechanisms, all of which result in increased activation of the pathway, as commonly seen in many cancer subtypes. Once PI3K signaling is activated, it can act on a diverse array of substrates including mTOR, a master regulator of protein translation. The PI3K/Akt/mTOR pathway is an attractive therapeutic target in cancer not only because it is the second most frequently altered pathway after p53, but also because it serves as a convergence point for many stimuli. Through its downstream substrates, this pathway controls key cellular processes, such as transcription, apoptosis, cell cycle progression, and translation ( Fig. 1 ).
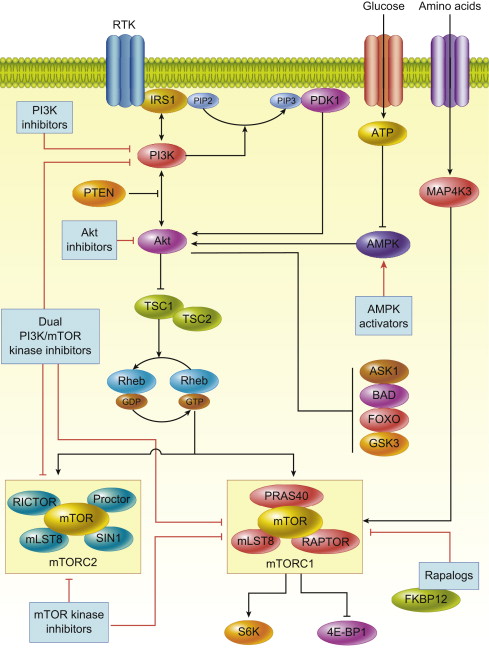
PI3 Kinase
PI3K signaling can be activated by multiple stimuli: activated tyrosine kinase growth factor receptors; cell adhesion molecules, such as integrins and G-protein-coupled receptors (GPCR); and oncogenes, such as Ras . PI3K is a member of the lipid kinase family that is divided into 3 classes, each differentially activated by stimuli. Class IA isoforms are activated by tyrosine kinase receptors or Ras ; class IB are activated by heterotrimeric G proteins or Ras ; class II PI3K isoforms are activated through insulin receptors, growth factor receptors, and integrins; and class III kinases are thought to be constitutively active. Despite all isoforms having unique lipid substrate specificity, all initiate downstream signaling by phosphorylating the D3 position of phosphatidylinositol rings at the cell membrane.
The class IA PI3K enzymes are the most relevant to activation of the PI3K/Akt/mTOR pathway, because they catalyze the generation of phosphatidylinositol-3, 4, 5-trisphosphate (PIP3) from phosphatidylinositol-4, 5-bisphosphate (PIP2). PI3K activation occurs through engagement of Src homology 2 (SH2) domains with phospho-tyrosine residues on activated growth factor receptors or through direct interaction with activated Ras .
PDK-1 and Akt are the crucial downstream kinases, and their activation depends on the generation of PIP3 and PIP2. These 3-phosphoinositides bind to the pleckstrin homology (PH) domains of PDK-1 and Akt to cause translocation of each kinase to the plasma membrane, where both are subsequently activated.
Akt
The phosphatase and tensin homolog deleted from chromosome 10 (PTEN), a tumor suppressor that dephosphorylates membrane phosphatidylinositols, is a key negative regulator of the effects of PI3K. Once PDK-1 is activated by PIP3, it propagates the signal to the serine/threonine kinase Akt by phosphorylating its catalytic domain. Akt has 3 isoforms (Akt1, 2, and 3), which are structurally similar and are expressed in most tissues. PDK-1 phosphorylates Akt1 in its activation loop on threonine 308 (T308), an event that alone stimulates partial activation of Akt. Full activation of Akt1 also requires phosphorylation at serine 473 (S473) in its regulatory domain. Phosphorylation of homologous residues in Akt2 and Akt3 occurs by the same mechanism. Several kinases are capable of phosphorylating Akt at S473, including PDK-1, integrin-linked kinase (ILK), an ILK-associated kinase, Akt itself, DNA-dependent protein kinase (DNA-PK), and mTORC2. Because many kinases are capable of S473 phosphorylation, this suggests that cell type–specific mechanisms of regulating Akt activity may exist or that different S473 kinases may be stimulated under different conditions.
Akt can be regulated by phosphorylation at other sites or by binding to other proteins in addition to phosphorylation at T308 and S473. For example, PKC-z, an isoform of protein kinase C, inhibits phosphorylation of Akt at T34 in the PH domain. Tyrosine (Y) phosphorylation at Y474 can also affect activation of Akt. Inositol polyphosphate 4-phosphatase type II (INPP4B), a tumor suppressor in human epithelial cells, is another inhibitor of PI3K/Akt signaling. In addition, S6 kinase 1 (S6K1), a downstream substrate of mTOR plays an important role in negative feedback regulation of Akt by catalyzing an inhibitory phosphorylation on insulin receptor substrate (IRS) proteins, abolishing their association and activation of PI3K, adding further complexity to the regulation of Akt kinase activity. In addition, Akt activity can also be modulated by Akt-binding proteins, such as heat shock protein 90, T-cell leukemia/lymphoma protein-1, carboxyterminal modulator protein, c-Jun N-terminal kinase (JNK)-interaction protein, and Tribbles homolog 3. Whether these mechanisms play an important role in cancer biology is not clearly known; however, the fact that multiple mechanisms of modulating Akt activity exist suggests that cell- and context-specific modes of regulation are involved; likewise, targeting these may lead to developments in PI3K/Akt pathway inhibitors.
Akt has numerous substrates that have been identified and validated through bioinformatics approaches. These substrates control key cellular processes, such as growth, including transcription, translation, cell cycle progression, and survival, including apoptosis, autophagy, and metabolism. With a few exceptions, Akt has an inhibitory effect on its multiple targets. However, as most Akt targets are negative regulators, the net result of Akt activation is cellular activation. For example, Akt phosphorylates forkhead box O1 (FoxO1) and other forkhead family members and results in inhibition of transcription of pro-apoptotic genes, such as Fas ligand, insulin-like growth factor binding protein 1 ( IGFBP1 ), and bisindolyl maleimide ( Bim ). Conversely, the inflammatory kinases (IKK), following Akt phosphorylation, increase NF- k B activity and the transcription of prosurvival genes. Akt, by phosphorylating and inactivating proapoptotic proteins, such as BCL2-associated agonist of cell death (Bad), directly regulates apoptotic machinery through Bad’s regulation and control of cytochrome c release from mitochondria. Akt is also involved in the regulation of apoptosis signal-regulating kinase-1 (ASK-1) and mitogen-activated protein kinase (MAPK) kinase, both of which are involved in stress and cytokine-induced cell death. Akt regulates cell cycle progression through the cyclin-dependent kinase inhibitors, p21WAF1/CIP1 and p27KIP1. Inhibition of glycogen synthase kinase-3beta (GSK-3beta) by Akt also stimulates cell cycle progression by stabilizing cyclin D1 expression. Akt also plays an important role in protein translation by phosphorylating tuberous sclerosis complex 2 (TSC2, also called tuberin) and mTOR. Thus, Akt inhibition can result in numerous effects on cancer cells that could contribute to an antitumor response.
mTOR
mTOR is an atypical serine/threonine protein kinase that belongs to the PI3K-related kinase family. mTOR exists in 2 multiprotein complexes: mTOR complex 1 and 2 (mTORC1 and mTORC2). mTORC1 is a complex of the mTOR protein, mammalian LST8, proline-rich Akt1 substrate 40 (PRAS40), and raptor, whereas mTORC2 consists of a complex of mTOR, mLST8, mSIN1, protor, and rictor. Akt activates mTOR through at least 2 mechanisms: either directly by phosphorylating mTORC1 at S2448 or indirectly through TSC2. TSC2 inactivation through phosphorylation by Akt results in upregulation of mTORC1 activity through a cascade of signaling molecules. A GTPase-activating domain of TSC2 catalyzes the conversion of the Ras-like protein, Ras homolog enriched in brain (Rheb)-GTP to Rheb-GDP leading to inactivation of mTOR function. Thus, Akt by decreasing TSC2 activity, increases levels of Rheb-GTP, which then leads to activation of mTORC1. mTORC1 plays a pivotal role in protein translation through its substrate: eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1). Hyperphosphorylation of 4E-BP1 by mTORC1, inhibits its binding to eukaryotic initiation factor 4E (eIF4E), thereby activating cap-dependent translation. In addition to protein translation, mTORC1 regulates cell proliferation, survival, and angiogenesis by regulating eIF4E-mediated translation of Bcl-2, Bcl-xL, and vascular endothelial growth factor (VEGF). mTORC1 also phosphorylates S6K1, which in turn leads to phosphorylation of S6 ribosomal protein, and other targets, including insulin receptor substrate 1 (IRS-1), eukaryotic initiation factor 4B (eIF4B), programmed cell death 4 (PDCD4), eukaryotic elongation factor-2 kinase (eEF2K), mTOR, and glycogen synthase kinase-3, which are implicated in cellular transformation. In addition, mTORC1 regulates the transition from G1 to S phase through downregulation of cyclin D1 and c-Myc, which are required for progression through this phase of the cell cycle.
In comparison with mTORC1, little is known about mTORC2 signaling. mTORC2 responds to growth factors, such as insulin, through a poorly defined mechanism that requires PI3K and ribosomes, as ribosomes are needed for mTORC2 activation and mTORC2 binds them in a PI3K-dependent fashion. mTORC2 controls several members of the AGC subfamily of kinases, including Akt, serum-induced and glucocorticoid-induced protein kinase 1 (SGK1), and protein kinase C-α (PKC-α). Akt is phosphorylated by mTORC2 at its hydrophobic motif (S473), a site required for its maximal activation. mTORC2 deletion, associated with defective Akt-Ser473 phosphorylation, impairs the phosphorylation of some Akt targets, including forkhead box O1/3a (FoxO1/3a), whereas other Akt targets, such as TSC2 and GSK-3beta, remain unaffected. mTORC2 also directly activates SGK1, a kinase controlling ion transport and growth. In contrast to Akt, mTORC2 deletion results in complete loss of SGK-1 activity. As SGK1 phosphorylates FoxO1/3a residues that are also phosphorylated by Akt, loss of SGK1 activity is probably responsible for the reduction in FoxO1/3a phosphorylation in mTORC2-depleted cells. PKC-α is another AGC kinase regulated by mTORC2. Along with other effectors, such as paxillin and Rho GTPases, mTORC2 plays a role in cell migration by regulating phosphorylation of PKC-α and control of the actin cytoskeleton in cell-type-specific fashion.
PI3K/Akt/mTOR signaling pathway
Initiation of signaling through the PI3K/Akt/mTOR pathway occurs through several mechanisms, all of which result in increased activation of the pathway, as commonly seen in many cancer subtypes. Once PI3K signaling is activated, it can act on a diverse array of substrates including mTOR, a master regulator of protein translation. The PI3K/Akt/mTOR pathway is an attractive therapeutic target in cancer not only because it is the second most frequently altered pathway after p53, but also because it serves as a convergence point for many stimuli. Through its downstream substrates, this pathway controls key cellular processes, such as transcription, apoptosis, cell cycle progression, and translation ( Fig. 1 ).
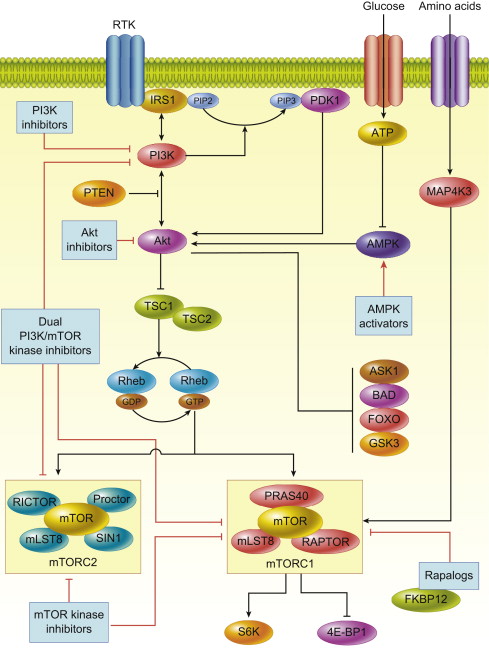
PI3 Kinase
PI3K signaling can be activated by multiple stimuli: activated tyrosine kinase growth factor receptors; cell adhesion molecules, such as integrins and G-protein-coupled receptors (GPCR); and oncogenes, such as Ras . PI3K is a member of the lipid kinase family that is divided into 3 classes, each differentially activated by stimuli. Class IA isoforms are activated by tyrosine kinase receptors or Ras ; class IB are activated by heterotrimeric G proteins or Ras ; class II PI3K isoforms are activated through insulin receptors, growth factor receptors, and integrins; and class III kinases are thought to be constitutively active. Despite all isoforms having unique lipid substrate specificity, all initiate downstream signaling by phosphorylating the D3 position of phosphatidylinositol rings at the cell membrane.
The class IA PI3K enzymes are the most relevant to activation of the PI3K/Akt/mTOR pathway, because they catalyze the generation of phosphatidylinositol-3, 4, 5-trisphosphate (PIP3) from phosphatidylinositol-4, 5-bisphosphate (PIP2). PI3K activation occurs through engagement of Src homology 2 (SH2) domains with phospho-tyrosine residues on activated growth factor receptors or through direct interaction with activated Ras .
PDK-1 and Akt are the crucial downstream kinases, and their activation depends on the generation of PIP3 and PIP2. These 3-phosphoinositides bind to the pleckstrin homology (PH) domains of PDK-1 and Akt to cause translocation of each kinase to the plasma membrane, where both are subsequently activated.
Akt
The phosphatase and tensin homolog deleted from chromosome 10 (PTEN), a tumor suppressor that dephosphorylates membrane phosphatidylinositols, is a key negative regulator of the effects of PI3K. Once PDK-1 is activated by PIP3, it propagates the signal to the serine/threonine kinase Akt by phosphorylating its catalytic domain. Akt has 3 isoforms (Akt1, 2, and 3), which are structurally similar and are expressed in most tissues. PDK-1 phosphorylates Akt1 in its activation loop on threonine 308 (T308), an event that alone stimulates partial activation of Akt. Full activation of Akt1 also requires phosphorylation at serine 473 (S473) in its regulatory domain. Phosphorylation of homologous residues in Akt2 and Akt3 occurs by the same mechanism. Several kinases are capable of phosphorylating Akt at S473, including PDK-1, integrin-linked kinase (ILK), an ILK-associated kinase, Akt itself, DNA-dependent protein kinase (DNA-PK), and mTORC2. Because many kinases are capable of S473 phosphorylation, this suggests that cell type–specific mechanisms of regulating Akt activity may exist or that different S473 kinases may be stimulated under different conditions.
Akt can be regulated by phosphorylation at other sites or by binding to other proteins in addition to phosphorylation at T308 and S473. For example, PKC-z, an isoform of protein kinase C, inhibits phosphorylation of Akt at T34 in the PH domain. Tyrosine (Y) phosphorylation at Y474 can also affect activation of Akt. Inositol polyphosphate 4-phosphatase type II (INPP4B), a tumor suppressor in human epithelial cells, is another inhibitor of PI3K/Akt signaling. In addition, S6 kinase 1 (S6K1), a downstream substrate of mTOR plays an important role in negative feedback regulation of Akt by catalyzing an inhibitory phosphorylation on insulin receptor substrate (IRS) proteins, abolishing their association and activation of PI3K, adding further complexity to the regulation of Akt kinase activity. In addition, Akt activity can also be modulated by Akt-binding proteins, such as heat shock protein 90, T-cell leukemia/lymphoma protein-1, carboxyterminal modulator protein, c-Jun N-terminal kinase (JNK)-interaction protein, and Tribbles homolog 3. Whether these mechanisms play an important role in cancer biology is not clearly known; however, the fact that multiple mechanisms of modulating Akt activity exist suggests that cell- and context-specific modes of regulation are involved; likewise, targeting these may lead to developments in PI3K/Akt pathway inhibitors.
Akt has numerous substrates that have been identified and validated through bioinformatics approaches. These substrates control key cellular processes, such as growth, including transcription, translation, cell cycle progression, and survival, including apoptosis, autophagy, and metabolism. With a few exceptions, Akt has an inhibitory effect on its multiple targets. However, as most Akt targets are negative regulators, the net result of Akt activation is cellular activation. For example, Akt phosphorylates forkhead box O1 (FoxO1) and other forkhead family members and results in inhibition of transcription of pro-apoptotic genes, such as Fas ligand, insulin-like growth factor binding protein 1 ( IGFBP1 ), and bisindolyl maleimide ( Bim ). Conversely, the inflammatory kinases (IKK), following Akt phosphorylation, increase NF- k B activity and the transcription of prosurvival genes. Akt, by phosphorylating and inactivating proapoptotic proteins, such as BCL2-associated agonist of cell death (Bad), directly regulates apoptotic machinery through Bad’s regulation and control of cytochrome c release from mitochondria. Akt is also involved in the regulation of apoptosis signal-regulating kinase-1 (ASK-1) and mitogen-activated protein kinase (MAPK) kinase, both of which are involved in stress and cytokine-induced cell death. Akt regulates cell cycle progression through the cyclin-dependent kinase inhibitors, p21WAF1/CIP1 and p27KIP1. Inhibition of glycogen synthase kinase-3beta (GSK-3beta) by Akt also stimulates cell cycle progression by stabilizing cyclin D1 expression. Akt also plays an important role in protein translation by phosphorylating tuberous sclerosis complex 2 (TSC2, also called tuberin) and mTOR. Thus, Akt inhibition can result in numerous effects on cancer cells that could contribute to an antitumor response.
mTOR
mTOR is an atypical serine/threonine protein kinase that belongs to the PI3K-related kinase family. mTOR exists in 2 multiprotein complexes: mTOR complex 1 and 2 (mTORC1 and mTORC2). mTORC1 is a complex of the mTOR protein, mammalian LST8, proline-rich Akt1 substrate 40 (PRAS40), and raptor, whereas mTORC2 consists of a complex of mTOR, mLST8, mSIN1, protor, and rictor. Akt activates mTOR through at least 2 mechanisms: either directly by phosphorylating mTORC1 at S2448 or indirectly through TSC2. TSC2 inactivation through phosphorylation by Akt results in upregulation of mTORC1 activity through a cascade of signaling molecules. A GTPase-activating domain of TSC2 catalyzes the conversion of the Ras-like protein, Ras homolog enriched in brain (Rheb)-GTP to Rheb-GDP leading to inactivation of mTOR function. Thus, Akt by decreasing TSC2 activity, increases levels of Rheb-GTP, which then leads to activation of mTORC1. mTORC1 plays a pivotal role in protein translation through its substrate: eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and S6 kinase 1 (S6K1). Hyperphosphorylation of 4E-BP1 by mTORC1, inhibits its binding to eukaryotic initiation factor 4E (eIF4E), thereby activating cap-dependent translation. In addition to protein translation, mTORC1 regulates cell proliferation, survival, and angiogenesis by regulating eIF4E-mediated translation of Bcl-2, Bcl-xL, and vascular endothelial growth factor (VEGF). mTORC1 also phosphorylates S6K1, which in turn leads to phosphorylation of S6 ribosomal protein, and other targets, including insulin receptor substrate 1 (IRS-1), eukaryotic initiation factor 4B (eIF4B), programmed cell death 4 (PDCD4), eukaryotic elongation factor-2 kinase (eEF2K), mTOR, and glycogen synthase kinase-3, which are implicated in cellular transformation. In addition, mTORC1 regulates the transition from G1 to S phase through downregulation of cyclin D1 and c-Myc, which are required for progression through this phase of the cell cycle.
In comparison with mTORC1, little is known about mTORC2 signaling. mTORC2 responds to growth factors, such as insulin, through a poorly defined mechanism that requires PI3K and ribosomes, as ribosomes are needed for mTORC2 activation and mTORC2 binds them in a PI3K-dependent fashion. mTORC2 controls several members of the AGC subfamily of kinases, including Akt, serum-induced and glucocorticoid-induced protein kinase 1 (SGK1), and protein kinase C-α (PKC-α). Akt is phosphorylated by mTORC2 at its hydrophobic motif (S473), a site required for its maximal activation. mTORC2 deletion, associated with defective Akt-Ser473 phosphorylation, impairs the phosphorylation of some Akt targets, including forkhead box O1/3a (FoxO1/3a), whereas other Akt targets, such as TSC2 and GSK-3beta, remain unaffected. mTORC2 also directly activates SGK1, a kinase controlling ion transport and growth. In contrast to Akt, mTORC2 deletion results in complete loss of SGK-1 activity. As SGK1 phosphorylates FoxO1/3a residues that are also phosphorylated by Akt, loss of SGK1 activity is probably responsible for the reduction in FoxO1/3a phosphorylation in mTORC2-depleted cells. PKC-α is another AGC kinase regulated by mTORC2. Along with other effectors, such as paxillin and Rho GTPases, mTORC2 plays a role in cell migration by regulating phosphorylation of PKC-α and control of the actin cytoskeleton in cell-type-specific fashion.
Pathogenesis of cancer by aberrations in the PI3K/Akt/mTOR pathway
Aberrations in the PI3K/Akt/mTOR pathway can occur through multiple mechanisms, resulting in pathway activation and contributing to the development of many human cancer types. Genomic aberrations affecting the PI3K pathway include germline and somatic mutations, amplifications, rearrangements, methylation, overexpression, and aberrant splicing, resulting in decreased expression or function of PTEN, amplification or mutation of PIK3CA , or amplification of Akt . The pathway is also triggered by activation of growth factor receptors, including human epidermal growth factor receptor 2 (HER2) and insulin-like growth factor receptor (IGFR), through autocrine growth loops, through mutations or overexpression of the growth factor receptors themselves, or by additional intracellular signaling molecules ( Table 1 ).
| Genetic Aberration | Tumor Type | Frequency, % |
|---|---|---|
| PTEN loss | Glioblastoma | 54–74 |
| Endometrial | 32–83 | |
| Gastric | 47 | |
| Prostate | 29 | |
| Breast | 39 | |
| Melanoma | 44–57 | |
| PTEN mutation | Glioblastoma | 17–44 |
| Endometrial | 36–50 | |
| Gastric | 7 | |
| Prostate | 12 | |
| Breast | 0–4 | |
| Melanoma | 7 | |
| PIK3CA amplification | Cervix | 69 |
| Gastric | 36 | |
| Lung (squamous) | 60 | |
| Head and neck | 37 | |
| Ovary | 25 | |
| PIK3CA mutation | Breast | 21–40 |
| Colorectal | 13–32 | |
| Glioblastoma | 5–8 | |
| Endometrial | 24–32 | |
| Hepatocellular | 6–36 | |
| Ovarian | 10 | |
| Gastric | 7 | |
| Akt amplification | Head and neck | 30 |
| Gastric | 20 | |
| Pancreas | 20 | |
| Ovary | 12 | |
| Akt1 mutation | Thyroid | 5 |
| Endometrial | 4 | |
| Breast | 4 | |
| Akt2 mutation | Breast | 0.8–1 |
| Akt3 mutation | Breast | 1 |
Loss of PTEN Function
PTEN is targeted by a number of regulatory events, many of which are aberrant in cancer, suggesting that PTEN is a critical regulator of pathway function. PTEN functions both as a protein and lipid phosphatase. Its tumor suppressive function is attributed to lipid phosphatases, as loss of lipid phosphatase function of PTEN results in increased PI3k/Akt/mTOR pathway activation. Loss of PTEN function and its activity in cancer occurs through multiple mechanisms, which include mutations, loss of heterozygosity, methylation, aberrant expression of regulatory microRNA, protein instability, and protein phosphorylation. PTEN is the second most frequently mutated tumor suppressor gene and, as shown in multiple studies, it is mutated or deleted in many human cancer types, including brain, bladder, breast, prostate, and endometrial cancers. Epigenetic silencing is seen in some tumor types, in which PTEN mutations are rare ; methylation of PTEN has been observed in 69% of non–small-cell lung cancer. Methylation of the promoter region in PTEN is also seen in endometrial and gastric cancer.
PIK3CA Amplification or Mutation
PI3K can be activated as a result of overexpression of structurally normal protein or from mutations in the catalytic p110 or regulatory p85 subunits. PIK3CA is the gene that encodes the p110a catalytic subunit and is overexpressed in 40% of ovarian and 50% of cervical cancers. In several cancer types, somatic mutations of this gene have been detected that result in increased kinase activity. Nonsynonymous mutations that encode the helical and kinase domains of the protein have been seen in 32% of colorectal cancers. In breast cancer, PIK3CA mutations have been observed in 21.4% of tumors. PIK3CA mutations have also been detected in 27% of glioblastomas and 25% of gastric cancers. Mutations in the regulatory subunit p85 have also been detected. For example, p65, a truncated version of p85, was isolated from a tumor cell line that has shown to cause constitutive activation of PI3K and cellular transformation. Moreover, a constitutively active p85 mutant, as a result of SH2 domain deletion, has been detected in colon and ovarian cancers. Notably, mutations, particularly in exons 9 and 20 of PIK3CA , encoding the helical and kinase domains respectively, activate Akt signaling in some models. However, PIK3CA mutations are not always associated with PI3K/Akt/mTOR pathway activation in vitro and were not associated with PI3K/Akt/mTOR pathway activation in breast cancers in The Cancer Genome Atlas. This suggests that the effect of PIK3CA mutations may also be cell context–dependent, and in certain cancer types, such as in breast cancer, other major regulators of the pathway may need to be considered.
Amplification of Akt
Amplification of Akt isoforms has been observed in some cancer subtypes. Akt1 amplification has been detected in gastric carcinomas, and amplification of Akt2 has been observed in 12% of ovarian cancers, 10% to 20% of pancreatic carcinomas, and a subset of pancreatic cancers. In breast cancer, amplification of either Akt1 or Akt2 has been reported. Akt3 amplification has been observed in prostate cancer and hormone-independent breast cancer cell lines. E17K mutation in the Pleckstrin homology (PH) protein domain of Akt has been identified. This mutation allows Akt1 recruitment to the cellular membrane independent of PI3K, conferring transforming activity. Sequencing based on mass spectroscopy showed mutations in Akt1-E17K in fewer than 2% of the breast tumors evaluated, and in none of the cell lines evaluated.
Activation of Growth Factor Receptors
The PI3K/Akt/mTOR pathway is also activated by cell surface growth factor receptors. Increased activation of growth factor receptors in cancer can occur via amplification, activating mutations, or the release of growth factors that stimulate their receptors in an autocrine fashion. The ERBB family of receptor tyrosine kinases is the most important family of growth factor receptors that is frequently activated in cancer cells. ERBB includes ERBB1/epidermal growth factor receptor (EGFR), HER2, HER3, and HER4. These receptors dimerize to activate the PI3K/Akt/mTOR pathway. HER2/HER3 heterodimers are especially considered to be strong activators of the PI3K/Akt pathway, whereas tumor cells with HER2 overexpression exhibit constitutive activation of the pathway. HER2 receptor mutations can also lead to constitutive activation of the pathway. EGFRvIII, a truncated version of EGFR, lacks the extracellular ligand-binding domain and is constitutively active, resulting in activation of the PI3K/Akt pathway. Mutations in the kinase domains of EGFR and HER2 , but not in HER3 or HER4 , result in pathway activation and have also been described in lung cancer. Aberrant autocrine growth loops can also result in increased pathway activation via activation of ERBB family members. For example, overexpression of 2 ligands of EGFR, transforming growth factor-a (TGF-a) and amphiregulin, is associated with activation of EGFR and the PI3K/Akt pathway.
Pathway Activation in Human Dysplastic Lesions
PI3K/Akt/mTOR pathway activation is an early event in the tumorigenesis of multiple cancers, as activation of the pathway has been described in multiple preneoplastic lesions. Hamartoma syndromes such as Cowden syndrome ( PTEN mutations), Peutz-Jeghers syndrome ( LKB1 mutations), tuberous sclerosis ( TSC1/2 mutations), neurofibromatosis ( NF1 mutations), and probably Birt-Hogg-Dubé ( BHD/Folliculin mutations) have increased activation of PI3K/Akt/mTOR pathway. Compared with a normal nevus, a dysplastic nevus has increased Akt activation, whereas in case of breast tissue, phosphorylation of Akt, mTOR, and 4E-BP1 increases progressively from normal breast epithelium to hyperplasia and from abnormal hyperplasia to tumor invasion.
PI3K/Akt/mTOR pathway targeted therapy
The PI3K/Akt/mTOR pathway holds multiple putative therapeutic targets. Because homeostasis of the pathway is tightly regulated, it is necessary to identify mechanistic feedback loops and cross-talk with other signaling cascades to anticipate mechanisms of adaptive response and acquired resistance and thereby to develop rational combination therapies. The PI3K/Akt/mTOR pathway can be targeted by a variety of approaches, including (1) targeting kinases that lead to activation of Akt, PI3K, and PDK-1; (2) directly targeting PI3K; (3) directly inhibiting Akt; (4) targeting downstream effectors of Akt, such as mTOR; or (5) combination approaches ( Table 2 ).
| Molecular Targets | Agent | Company | Phase |
|---|---|---|---|
| PI3K inhibitors | BAY80-6946 | Bayer (Pitssburg, PA) | I/II |
| BKM120 | Novartis (New York, NY) | II/III | |
| GDC-0941 | Genentech (San Francisco, CA) | II | |
| PX866 | Oncothyreon (Seattle, WA) | I/II | |
| XL147 | Exelixis (San Francisco, CA) | I/II | |
| PIK3Cd | CAL-101 | Calistoga/Gilead (Seattle, WA) | III |
| PI3Kα | BYL719 | Novartis | I/II |
| MLN1117 | Millennium (Cambridge, MA) | I | |
| PDK-1 inhibitors | OSU-03012 (AR-12) | Arno (Flemington, MA) | I |
| UCN-01 | Keryx (New York, NY) | I/II | |
| Dual PI3K/mTOR inhibitors | BEZ235 | Novartis | I/II |
| GDC-0980 | Genentech | I/II | |
| PF-04691502 | Pfizer (New York, NY) | I/II | |
| PF-05212384 | Pfizer | I/II | |
| XL765 | Exelixis | I/II | |
| Multimodal inhibitor (PI3K, mTOR, DNA-PK, HIF-1α) | SF1126 | Semafore (Westfield, IN) | I |
| Akt inhibitors | AZD5363 | AstraZeneca (Wilmington, DE) | I |
| MK-2206 | Merck (Whitehouse Station, NJ) | I/II | |
| GSK2110183 | GlaxoSmithKline (Philadelphia, PA) | I | |
| Perifosine | Keryx | III | |
| mTOR kinase inhibitors | AZD2014 | AstraZeneca | I/II |
| AZD8055 | AstraZeneca | I | |
| MLN0128 | Millennium | I | |
| OSI-027 | OSI Oncology/Astellas (Long Island, NY) | I | |
| Rapalogs | Everolimus (RAD001) | Novartis | IV/FDA approved for breast cancer, RCC, PNET, subependymal giant cell astrocytoma (in TSC) |
| Ridaforolimus (MK8669) | ARIAD (Cambridge, MA)/MERCK | II | |
| Sirolimus | Wyeth/Pfizer (New York, NY) | III | |
| Temsirolimus (CCI779) | Wyeth/Pfizer | III/FDA approved for RCC |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


