This article summarizes data on translational studies to target the p53 pathway in cancer. It describes the functions of the p53 and Mdm-2 signaling pathways, and discusses current therapeutic approaches to target p53 pathways, including reactivation of p53. In addition, direct interaction and colocalization of the p53 and focal adhesion kinase proteins in cancer cells have been demonstrated, and different approaches to target this interaction are reviewed. This is a broad review of p53 function as it relates to the diagnosis and treatment of a wide range of cancers.
Key points
- •
The p53 gene is a tumor-suppressor gene whose genomic sequence contains 11 exons and spans 20 kilobases.
- •
p53 is the most commonly mutated gene in tumors, with up to 50% mutations in different types of cancers and 75% mutations in invasive cancers.
- •
Focal Adhesion Kinase (FAK) can suppress the transcriptional activity of p53 through its interaction.
- •
Understanding FAK biology during tumorigenesis, the mechanisms of its upregulation in different tumors, its role in stem cell biology, angiogenesis, and motility, and the mechanisms of its direct physical interaction with p53 protein and their downstream signaling pathways will be critical in developing targeted therapeutics.
Introduction
p53 is a guardian of the genome, and a tumor-suppressor gene that controls genetic stability and prevents cancer. p53 is highly mutated in different types of tumors or is inactivated by different mechanisms such as interacting with Mdm-2, Mdmx, Focal Adhesion Kinase (FAK), and other proteins. This review discusses different approaches to reactivate the p53 pathway either through targeting its inhibiting protein interactions with Mdm-2 and FAK proteins, through changing protein conformations with small-molecule inhibitors, through the introduction of wild-type p53, or through other pathways, focusing on future directions and perspectives in translational research on developing p53-targeting therapeutics. There are several homologous proteins to p53, such as p63 and p73, with similar and unique functions that are not discussed in this article.
Introduction
p53 is a guardian of the genome, and a tumor-suppressor gene that controls genetic stability and prevents cancer. p53 is highly mutated in different types of tumors or is inactivated by different mechanisms such as interacting with Mdm-2, Mdmx, Focal Adhesion Kinase (FAK), and other proteins. This review discusses different approaches to reactivate the p53 pathway either through targeting its inhibiting protein interactions with Mdm-2 and FAK proteins, through changing protein conformations with small-molecule inhibitors, through the introduction of wild-type p53, or through other pathways, focusing on future directions and perspectives in translational research on developing p53-targeting therapeutics. There are several homologous proteins to p53, such as p63 and p73, with similar and unique functions that are not discussed in this article.
p53 protein structure and function
p53 is a tumor-suppressor gene whose genomic sequence contains 11 exons and spans 20 kilobases; it is located at the short arm of the chromosome 17p13.1 region and encodes p53 protein. The p53 protein is a transcription factor that binds as a tetramer to the DNA consensus sequence 5′Pu-Pu-Pu-C-A/T-T/A-G-Py-Py-Py3′ (Pu, purine; Py, pyrimidine) in the promoters of the several genes, such as p21, Bax, Mdm-2, and PUMA, and activates their transcription. It has also been shown that p53 can repress transcription of several important genes such as survivin, Bcl-2, FAK, and others. The promoter of p53 does not contain a TATA box and consists of various binding sites for known transcription factors, such as NF-κB, Sp1, or c-Jun. The p53 gene encodes a 393-amino-acid tumor-suppressor protein, which contains several functional domains: 2 N-terminal transcriptional activating domains, TAD1 (amino acids [aa] 1–42) and TAD2 (aa 43–65); a proline-rich domain (aa 64–92); a central DNA-binding domain (aa 102–292); and a C-terminal tetramerization domain (aa 325–393) and basic region (aa 364–393) ( Fig. 1 ). The nuclear localization signal is located after the DNA-binding domain (aa 305–322) (see Fig. 1 ).
N-Terminal Transactivation Domain of p53
The 17 to 28 amino acids of the first N-terminal transactivation domain interact with Mdm-2 protein, double minute-2 homologue, which plays a major role in p53 degradation via the ubiquitin-proteasome pathway. p53 activates Mdm-2 transcription through binding to its promoter, and upregulates Mdm-2 expression. In turn, Mdm-2 binds to p53 and negatively regulates its stability and activity. Thus, Mdm-2 regulates p53 via an autoregulatory feedback loop in which both proteins control its cellular expression. Amino acids 22 to 26 are involved in the binding of histone acetyltransferase P300 (reviewed in Ref. ). The second transactivation domain (aa 43–63) activates transcription of several p53 transcriptional targets.
Proline-Rich Domain of p53
A proline-rich domain (aa 64–92) contains 5 PXXP motifs, is important for interaction with various proteins, and regulates apoptosis. The PXXP motifs of this domain were shown to create a binding site for Src-homology SH3 domains. A mutant deleted for the proline-rich domain in human p53 (Δ62–91) was not able to cause apoptosis but was able to maintain cell-cycle arrest. A polymorphism at the amino acid 72 of either proline or arginine could cause differences in binding of transcription factors, survival/apoptotic signaling, and response to chemotherapy agents.
Central DNA-Binding Domain
The core central region of p53 is highly conserved, homologous to the regions of other p53 family members (p63 and p73). The DNA-binding domain binds to promoter binding sites 5′Pu-Pu-Pu-C-A/T-T/A-G-Py-Py-Py3′. Several p53 mutation hot spots are located in this domain, such as: Arg175, Gly245, Arg248, Arg273, and Arg282 (see Fig. 1 ).
C-terminal Tetramerization Domain
This domain is involved in protein tetramerization and regulation of p53 activity. It contains the tetramerization region (aa 323–356) and a negative regulatory region (aa 363–393) (see Ref. for a review).
p53 mutations in cancer cells
p53 is the most commonly mutated gene in tumors, with up to 50% mutations in different types of cancers and 75% mutations in invasive cancers. Most mutations are missense mutations, with 4 of 5 mutations in the DNA-binding domain, only 1% in the N-terminal domain, and 4% in the C-terminal tetramerization domain (reviewed in Ref. ). The most frequent hot-spot mutations are: 175 Arg → His (breaks bond between L2 and L3 loop); 248 Arg → Gln (Trp) (breaks contact with DNA in minor groove); 273 Arg → His (Cys) (breaks contact with DNA in major groove); and 282 Arg → Trp (destabilizes H2 helix and DNA binding in the major groove and breaks contacts on the β-hairpin) (reviewed in Ref. ) Arginines 248 and 273 are involved in interaction of p53 with DNA, and arginines 175 and 282 stabilize the DNA-binding sequence of p53. Wild-type p53 binds to gene promoters with different affinity; for example, p53 activates p21 promoter with a higher affinity than the Bax promoter.
p53 function
p53 was discovered in 1979 and was first thought to play the role of oncogene, but was then revisited and found to be a tumor-suppressor gene. The model of the main functions of p53 is shown in Fig. 2 and is discussed in a recent review summarizing the first 30 years of p53 research. It is known that p53 is mutated in almost 50% of all tumors. Inactivation of the p53 gene is a critical step in tumorigenesis. Following induction by a variety of cell stresses, such as DNA damage, hypoxia, or the presence of activated oncogenes, p53 upregulates a set of genes that can promote either cell death, apoptosis, senescence, or growth arrest, such as p21 , GADD45 , cyclin G , and Bax , reviewed in Ref. Recently it was shown that p53 can repress promoter activities of several antiapoptotic genes and cell cycle genes (survivin, cyclin B1, cdc2, cdc25 c, stathmin, Map4, and bcl-2 ). Overexpression of p53-induced apoptosis and p53 inactivation caused a decrease in radiation-induced apoptosis. More recently, new functions of p53, such as metabolism and antioxidant defense, have been reported and reviewed (see Fig. 2 ).
p53 Binds Focal Adhesion Kinase Promoter and Represses its Activity
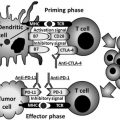
FAK is a 125-kDa nonreceptor kinase that plays an important role in survival signaling, adhesion, motility, invasion, angiogenesis, and proliferation. FAK was shown to be overexpressed in many types of tumors and has been proposed as a therapeutic target.
The first report on the indirect link of FAK and p53 signaling in apoptosis was reported by Ilic and colleagues, who demonstrated that FAK suppressed a p53-regulated apoptotic pathway in anchorage-dependent cells such as fibroblasts and endothelial cells. These investigators showed that in the absence of FAK function, p53-regulated apoptosis was activated by protein kinase C and phospholipase A 2 and that this process was inhibited by dominant-negative p53 and Bcl-2. However, these processes were not studied in tumors and cancer cell lines. In addition, immunohistochemical analysis of 115 endometrial carcinoma samples demonstrated a correlation between FAK and p53 overexpression.
The authors’ group cloned the FAK promoter and found 2 p53 binding sites in this area, and showed that p53 binds to the FAK promoter and inhibits its transcriptional activity. Moreover, mutant p53 with hot-spot mutations did not inhibit FAK promoter activity, whereas the wild type did. The global analysis of p53 transcription factor binding sites demonstrated that induction of HCT116 colon cancer cells with 5-fluorouracil downregulated FAK. It was therefore suggested that p53 can suppress metastasis through downregulation of metastasis-linked genes such as FAK. Because p53 is often mutated in cancer, the authors performed a population-based study on 600 tumors from patients with breast cancer, and found that p53 mutations highly correlated with FAK overexpression. Recently, another group found a high correlation between high FAK expression and low levels of p53 in oral squamous cell carcinoma, which may be explained by the absence of a repressive function on FAK promoter in p53-negative samples.
Direct p53 and FAK Protein Binding and Their Feedback Regulation
After finding that p53 regulates FAK promoter activity, the authors demonstrated that the N-terminal transactivation domain of p53 is physically and directly associated with the N-terminal domain of FAK. There have been several reports on the localization and function of the N-terminal part of FAK in the nucleus. The N-terminal domain of FAK caused apoptosis in breast-cancer cell lines, and its nuclear localization was regulated by caspase inhibitors in endothelial cells. p53 was also reported to be localized in the cytoplasm. p53 directly activated Bax and released proapoptotic molecules, activating multidomain protein complexes in the cytoplasm. This mechanism required the 62- to 91-amono acid proline-rich domain N-terminal domain of p53. The authors also showed that FAK binds the 7 amino acids (aa 65–71) in the proline region of p53. In addition, through computer modeling it was found that p53 peptides containing the amino acids 65 to 71 reactivated p53. This p53 peptide was modeled into the 3-dimensional structure of FAK, and small molecules that docked into this site of interaction were screened. In this way the authors identified novel small molecules targeting the FAK and p53 interaction that were able to activate p53 (see later discussion).
The authors have shown that FAK can suppress the transcriptional activity of p53 through its interaction, as p53-mediated activation of the p53 targets p21, Mdm-2, and Bax was blocked by overexpression of FAK, but not by ΔFAK-NT with a deleted N-terminal domain that binds p53. Thus, p53 regulates FAK and, in turn, FAK regulates p53. Thus FAK and p53 can be regulated through a feedback mechanism.
The binding of FAK and p53 was also studied by another group, who showed interaction of FAK with p53 and also with Mdm-2 proteins whereby binding of FAK to Mdm-2 facilitated p53 ubiquitination and degradation. The link between FAK and p53 was supported by a report on the FAK-interacting protein, FIP200. FIP200 overexpression resulted in increased p21 levels and caused growth arrest, and FIP200 interacted with FAK and p53. The investigators proposed a model of FAK-p53-FIP200 interaction, whereby FIP200/FAK and FIP200/p53 binding had competition and when FAK had low expression, FIP200 could bind p53 and increase its expression.
Recently, the authors showed that the stem-cell marker and transcription factor Nanog, which maintains stem-cell renewal and differentiation, was able to bind FAK promoter and upregulate its expression. In addition, FAK phosphorylated Nanog. Of note, Nanog is repressed by p53 that induced differentiation of embryonic stem cells. FAK is also repressed by p53, whereas it is activated is by Nanog. The mechanism of Nanog and FAK regulation and its cross-talk with p53 in cancer and stem cells requires further study. The novel mechanisms of FAK, p53, and Nanog survival function during carcinogenesis remain to be discovered.
p53-Targeted therapy
This section discusses therapeutic approaches to target p53 function, beginning with p53-targeted therapy. The discussion then focuses on FAK-p53-targeted therapy based on data on FAK and p53 interaction.
Translational approaches that target p53 for cancer treatment include therapies targeting mutant p53 and therapies targeting wild-type p53 (reviewed in Ref. ), and are shown in Fig. 3 .
Mutant p53–Targeted Therapy
To target mutant type p53, 2 therapeutic approaches are used: (1) deliver wild-type p53 with viruses (retroviruses or adenoviruses); and (2) use oncolytic therapy to destroy tumor cells with deleted or mutated p53 by lysis.
Exogenous expression of wild-type p53
Viral p53 delivery
Retroviruses are attractive agents for cancer gene therapy, as they integrate in the stable form into the genome and require cell division for transduction. The second strategy is to use adenoviruses, which are double-stranded DNA viruses. In contrast to retroviruses, their effect is not limited to highly proliferating cells and there is no risk of insertional mutagenesis or tumorigenesis.
Advexin
One of the adenoviral drugs is Ad5CMV-p53 or (Advexin, developed by Introgen Therapeutics Inc ; or Gendicine, Shenzden SiBiono Gene Tech ), which is a recombinant E1-deleted serotype 5 adenoviral vector encoding p53. In vitro and in vivo studies confirmed the therapeutic effect of Ad5CMVp53. This therapeutic approach was used effectively in phase I and II clinical trials, although some cases of tumor resistance were also demonstrated (reviewed in Ref. ). The drug was not approved by the Food and Drug Administration (FDA), although Gendicine (adenoviral p53) is used for the treatment of head and neck cancer in China and is effective in the treatment of hepatocellular carcinoma.
Oncolytic viruses
The second viral approach is to apply oncolytic viruses to kill p53-defective tumor cells, including the use of E1B-defective adenoviruses, which is a major approach.
Onyx-015
The delta 1520 virus (dl1520; ONYX-015), developed by Onyx Phramaceuticals, is an adenovirus that contains a deletion in the E1B region. ONYX-015 can be used as a selective drug against tumors with mutant p53. The problem of this and other adenoviral vectors is hepatotoxicity, and the FDA did not approve Onyx-015 after its use in numerous clinical trials.
Reactivation of mutant p53 with small molecules
To reactivate mutant p53, compounds were developed that targeted mutant p53 and reactivated wild-type p53 based on structural studies of p53 conformation, function, and regulation (see Fig. 3 ). The structures of all compounds are shown in review, and the compounds are presented here in chronologic order, according to when they were first reported (see Fig. 3 ).
Ellipticine (1998)
One of the first compounds that targeted mutant p53 was ellipticine, an alkaloid that had cytotoxic and antiproliferative activity in a panel of cell lines with mutant p53. Its derivative, 9-hydroxy-ellipticine (9HE), was developed in 1999. Later studies showed the off-target effects of ellipticine, such as targeting topoisomerase II, FOX, and other mechanisms.
CP31398 (1999)
In 1999, compound CP31398 (styrylquinazoline), which stabilized the DNA-binding domain of p53, was identified by high-throughput screening of a library of more than 100,000 synthetic small-molecule compounds by scientists from Pfizer Inc. The compound increased conformationally active p53 in cells with mutant p53, increased p53 transcriptional activity with p21 target in cells with mutant p53, and decreased tumor growth. CP31398 caused apoptosis in different cancer cell lines and stabilized wild-type p53 protein independently from Mdm-2. However, different quantitative biophysical methods under a wide range of conditions did not detect the interaction of CP31398 and p53 core domain, but detected intercalation of CP31398 with DNA and destabilization of DNA-p53 complex. The compound had several p53-independent functions, such as increase of apoptotic Bax protein. CP31398 did not affect binding of wild-type p53 with DNA and did not affect p63 and p73 homologues binding to DNA, but increased the amount and affinity ( K d ) of mutant p53 with DNA. CP31398 restored the functions of mutant p53 to suppress ultraviolet B–induced skin carcinogenesis in mice, which was associated with increased p53, p21, and BclXs. CP31398 was shown to have a chemopreventive effect on the development of intestinal adenoma in an animal model of familial adenomatous polyposis. The compound suppressed development of tumors by 36% and 75% at low and high doses, respectively, and increased expression of p53, p21, cleaved caspase-3, and PARP-1. The chemopreventive effect of CP31398 was observed in combination with celecoxib on azoxymethane-induced aberrant crypt foci and colon adenocarcinomas in F344 rats. The combination of CP31398 and celecoxib reduced the incidence of adenocarcinoma by 78% and its multiplicity by 90%. Rats fed with a combination of these drugs had increased p21 expression and apoptosis, and reduced tumor-cell proliferation.
PRIMA-1 (2002)
Another p53-reactivating compound was found in 2002 and named PRIMA (p53 Reactivation and Induction of Massive Apoptosis). PRIMA had antitumor activity in vivo in a mutant p53–dependent manner. It restored the p53-binding activity of R273H mutant and also other p53 mutants in the DNA-binding domain. A methylated form of PRIMA-1 was more active than the parent compound and synergized with cisplatin to induce tumor-cell apoptosis, and induced apoptotic Bax and PUMA p53 targets. PRIMA-1MET had suppressive effects in different mutant p53–carrying mouse tumors: mouse sarcomas, mammary carcinoma, and chemically induced fibrosarcoma. PRIMA-1 entered into phase I clinical trials based on its high efficacy in the animal models.
SCH529074 (2004)
Another small-molecule compound was discovered in 2004 and named SCH529074. This compound was able to bind the DNA-binding domain of p53, restore the growth-suppressive functions of mutant p53, and interrupt the HDM2-regulated ubiquitination of wild-type p53.
MIRA-3 (2005)
The MIRA-1 derivative, MIRA-3, was also able to reactivate mutant p53 and to express antitumor activity in tumor xenografts with human mutant p53 in vivo .
STIMA-1 (2008)
The compound STIMA-1 had structural similarities with CP-31398 and was able to reactivate mutant p53.
RETRA (2008)
Screening of a small-molecule library to induce p53-dependent reporter activity in A431 cells with R273H mutation identified another compound, RETRA-1 (REactivation of Transcriptional Reporter Activity). RETRA-1 showed antitumor activity in mouse xenografts, and also targeted homologous p53 protein p73. RETRA was suggested to sequester and release p73 from its inhibitory complex with mutant p53. Thus, targeting homologous to p53 proteins, such as p63 and p73, is a potential approach that can be used in therapy.
NSC319726 (2012)
In 2012 3 new compounds, NSC319725, NSC319726, and NSC328784, were found by in silico modeling screening of an National Cancer Institute (NCI) library of small-molecule compounds in a panel of 60 cancer cell lines carrying p53 mutations, and were reported to selectively kill cancer cells while not affecting normal cells. After compound validation, NSC319726 had the best and most specific killing effects on cancer cells, changing the conformation of p53 and restoring wild-type function of p53R175 mutant. The compound killed R172Hp53 knock-in mice, caused apoptosis, and activated p21 (p53 target) in mice xenograft tumors in a 175-allele–specific mutant p53–dependent manner. The activity of the compound was dependent on the zinc-chelating properties as well as redox changes. It was concluded that the in silico method on a panel of cancer cell lines promises to be a future strategy in searching for p53-reactivating compounds.
Wild-Type p53–Targeted Therapy
The small-molecule compounds were described to mostly target the p53 and Mdm-2 interaction, and are described herein (see Fig. 3 ). Also described are the FAK-p53 pathway small-molecule inhibitors developed by the authors, along with analysis of the structural data of these complexes and the small molecules targeting these complexes.
p53 and Mdm-2 interaction
The p53–Mdm-2 complex does not involve large protein areas, as many protein-protein interactions make it suitable for the development of targeted drug therapy. Of the 6 p53 amino acids from the p53 N-terminal transactivation domain, 18 TFSDLW 23 specifically binds to the pocket in the Mdm-2 protein. The cocrystallization of p53 peptide and Mdm-2 protein identified the 109-amino-acid region of the amino-terminal domain of Mdm-2 bound to p53 peptide and detected that Phe19, Trp23, and Leu26 were deeply inserted inside the hydrophobic Mdm-2 cleft. Two hybrid genetic analyses identified amino acids of Mdm-2 involved in interaction with p53, and included G58, D68, V75, and C77 amino acids of the N-terminal domain of Mdm-2. A later study identified additional sites in the N-terminal transactivation domain of p53, residues 40 to 45 and residues 49 to 54, which are involved in binding with Mdm-2. This binding is weaker than at the main site of p53–Mdm-2 interaction, but can provide additional approaches to drug development. The first drugs developed to target p53-Mdm-2 interaction were peptides, which were superseded by small-molecule compounds, owing to their higher stability, bioavailability, and other pharmacologic and pharmacokinetic properties (see later discussion).
Sulfonamide (2004)
Sulfonamide I was identified by computational screening using a 3-dimensional pharmacophore model and small molecules from the National Center for Biotechnology Information database, and this compound effectively inhibited p53–Mdm-2 interaction and activated p53. In the binding assay IC 50 was 32 μM, which was higher than that of p53 peptide, which caused 100% inhibition of p53–Mdm-2 binding at 100 μM. The functional efficacy was demonstrated in a reporter luciferase assay, whereby sulfonamide increased p53-transcriptional activity in Mdm-2-overexpressing cell line.
Nutlins (2004)
The small-molecule drugs that target p53 and Mdm-2 interaction were developed by La Roche in Nutley, New Jersey and were named Nutlins. These compounds bind Mdm-2 in the p53-binding pocket with a nanomolar range of binding affinity. The first 3 compounds, Nutlin-1, -2, and -3, share a common structure. The binding of Nutlin with Mdm-2 was confirmed by cocrystallization studies. The compounds activated the p53 pathway in cancer cells, activated the p21 target, caused apoptosis, and led to inhibition of tumor xenograft growth. The Nutlin-3 antitumor activity was shown in different types of cancer, including acute myeloid leukemia, neuroblastoma, osteosarcoma, and prostate cancer. The effect of Nutlins was p53 dependent. An oral Nutlin-3 entered clinical trials recently for the treatment of patients with solid tumors and hematologic cancers.
RITA (2004)
RITA (Reactivation of p53 and Induction of Tumor Apoptosis) was identified by screening a chemical library from the NCI set of compounds. RITA inhibited growth of colon cancer cell HCT116 more efficiently than p53−/− cells and caused a p53-dependent decrease of xenograft tumor growth in vivo . RITA prevented interaction of p53 and Mdm-2 in vitro , owing to p53 conformational change. RITA reactivated p53 and induced apoptosis in multiple myeloma cancer and leukemia cells. RITA synergized with Nutlin to induce apoptosis in melanoma. RITA reactivated p53 and also activated the JNK-1 pathway in multiple myeloma cancer cells. RITA caused a decrease of cervical carcinoma xenograft growth in vivo , associated with reactivation with p53 transcriptional targets: Bax, Noxa-1, and PUMA-1.
Terphenyl 14 (2005)
The computational screening of a small terphenyl library identified terphenyl 14, which was able to bind to the hydrophobic groove of Mdm-2 protein, to mimic the α-helical region of p53, and to disrupt p53–Mdm-2 protein interaction.
Benzodiazepinediones (2005)
High-throughput screening of chemical libraries using the assay that detected the effect of compounds on protein stability identified the benzodiazepinedione family of compounds, and the lead compound I stabilized p53 and induced p21 expression. The optimization of compound I resulted in TDP521252 and TD665759, which both inhibited Mdm-2. TDP521252 and TDP665759 inhibited the proliferation of wild-type p53 cell lines with an IC 48 of 14 and 0.7 μM, respectively, and disrupted the complex of Mdm-2 and p53. Administration of TD665759 to mice increased p21 in liver samples. The compound synergized with doxorubicin in an A375 xenograft model, and decreased tumor growth.
Quinolinole (NSC66811) (2006)
The computational screening of small molecules identified 354 Mdm-2 potential candidates to disrupt interaction with p53. The binding assay demonstrated that quinolinole (7-[anilino(phenyl)methyl]-2-methyl-8-quinolinol) (NSC66811) is the best to inhibit Mdm-2 in nanomolar concentrations, with a K i (120 nM), was lower than that of p53 peptide. The compound mimicked 3 amino acids of p53 that were involved in interaction with Mdm-2, and disrupted interaction with Mdm-2.
MI (2006)
The MI compounds were generated by the University of Michigan and Ascenta, and belong to the family of potent tryptophan-based inhibitors, which bind to Mdm-2 with high affinity, mimicking the binding of Trp23 from the p53 peptide, and disrupt the interaction of Mdm-2 with p53. At first oxindole was found to mimic the interaction of Trp23. The subsequent chemical modifications and optimizations resulted in a series of MI compounds. One of the most potent inhibitors was MI-63, with a K i of 3 nM, IC 50 of 280 nM, and induced p53 and p21 expression. The MI-43 was designed to target 4 amino acids of p53 peptide: Phe19, Trp23, Leu22, and Leu26. MI-43 inhibited cell growth and survival of colon cancer cells with wild-type p53. The MI-43 compound induced cell-cycle arrest that required p53 and p21. The optimization of MI-63 resulted in MI-219, inhibiting tumor growth in vivo . The next derivative of MI-219 was MI-319, which demonstrated efficient tumor growth suppression of lymphoma cells. MI-319 synergized with cisplatin and decreased cell growth in pancreatic cancer.
Isoindolinones (2006)
Isoindolinones were identified as scaffold Mdm-2 inhibitors, blocking the pocket of interaction with p53. The IC 50 values of these inhibitors are 5 to 16 μM in growth inhibition assays and reporter assays, and they inhibit the binding of Mdm-2 to p53. The most potent inhibitor was (+)- R- 3-(4-chlorophenyl)-3-(1-hydroxymethylcyclopropylmethoxy)-2-(4-nitrobenzyl)-2,3-dihydroisoindol-1-one or isoindolinone compound 74a, which effectively disrupted interaction of Mdm-2 and p53 and activated p53, p21, and Mdm-2 transcription. The IC 50 of 74a was 170 nM, versus 61 nM of Nutlin, in an Mdm-2 binding assay, and was similar in viability assays. The cellular activity of 74a was p53 dependent.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


