Fig. 20.1
Harnessing dendritic cells and T cells in the tumor microenvironment: efficacy and limitations
20.3.4 Exogenous and Endogenous Dendritic Cell Engineering
- 1.
Exogenous approaches: One approach to enhancing cytotoxic T-cell activity against tumors is the harnessing of dendritic cells through exogenous approaches. In this manner, purifying DCs from the patient and using them to boost sensitization of adoptive T cells to tumor antigens (thus presented optimally through MHC on the DCs) ex vivo may allow reinfusion of a highly efficacious population of antitumor T cells back into the patient in the appropriate clinical laboratory platform [31]. This is a strategy that has shown efficacy in mouse models, although thus far for human carcinoma, its most successful use has been in metastatic prostate carcinoma, employing peripheral blood mononuclear cells (PBMCs) along with delivery of antigen (prostatic acid phosphatase) and an adjuvant cytokine (GM-CSF) to facilitate DC maturation and effector functions prior to reinfusion back into the patient [53]. It has been difficult to demonstrate consistent durable antitumor effects, however, with exogenous DC-based approaches. One major challenge (even if the DC is conditioned very efficiently, with successful achievement of a strong antitumor phenotype ex vivo) is the possibility of rapid alteration in DC behavior and DC expression upon reentry into a host. Moreover, when ideal “tumor antigens” are paired with DCs ex vivo in such approaches, not all antigens are presented efficiently in the context of MHC-I; and so the downstream induction of cytotoxic T-cell responses may still be very limited [54].
- 2.
Endogenous DC engineering: This is a concept that might be considered broadly as one that modulates DC behavior through strategies that do not involve isolation of DCs outside of the host. Of course, vaccines would technically be considered as one form of “endogenous” DC modulation [23], although we would reserve the word “engineering” for non-antigen approaches to specifically modulate DC behavior in a way that improves antitumor responses (where the DC employs “natural” endogenous tumor antigens to induce T-cell responses). Our own laboratory has interest in this approach, from a glycobiology standpoint: An example of such engineering in this context might involve small-molecule inhibition of a key glycan co-receptor involved in driving immature DC traffic and regulating or inhibiting DC maturation responses. Indeed, we have found that DCs induced by certain cytokines such as TNF-α strongly upregulate the heparan sulfate proteoglycan syndecan-4. Silencing of this molecule or the sulfation of its glycan chains (which facilitate the actions of chemokines that drive DC traffic) appears to slow DC traffic [55], which may allow for greater cross presentation of tumor antigens to T cells in tumor-draining lymphoid organs (e.g., tumor DLNs). Glycan mutations in such DCs also appear to strikingly increase DC maturation [55]. Figure 20.2 illustrates how targeting a major proteoglycan on DCs might increase maturation while inhibiting the rate of trafficking of immature DCs in the tumor and draining lymph nodes, ultimately resulting in improvements in T-cell immunity that result in tumor growth inhibition. While this might also impact activated DCs outside of the tumor microenvironment (not necessarily in a detrimental manner, however), it would serve as a promising way to endogenously alter DCs that uniquely express this proteoglycan in the tumor and DLN microenvironments.
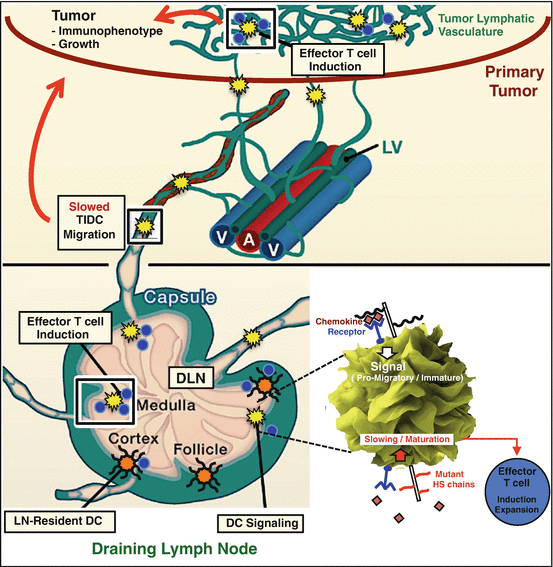
Fig. 20.2
Example of a novel glycan-targeting approach to augment antitumor immunity through endogenous dendritic cell functions: genetic reduction in the sulfation of glycans expressed on the surface of dendritic cells (shown in yellow) that traffic within the tumor microenvironment may alter their kinetics through slowing of glycan-mediated chemokine-dependent trafficking of immature dendritic cells from tumor to draining lymph node, with mechanisms illustrated in “magnified” dendritic cell shown within the inset at lower right. In particular, the altered glycan heparan sulfate (HS), targeted through mutation in this case, may be associated with a phenotype of DC slowing and increased maturation (lower right). This targeting may also increase maturation of tumor-associated dendritic cells in a manner that promotes effector T-cell functions following efficient tumor antigen presentation. Further work is needed to elucidate mechanisms for the latter; however, the ultimate result may contribute to an antitumor immune phenotype [55] and inhibited tumor growth (red arrows , implying inhibition). This illustrates one of possibly several novel ways to endogenously augment dendritic cell functions in the lung carcinoma and its draining lymph nodes (bottom); illustration and zoom of DC modified from Refs. [56, 57]
20.4 Conclusions and Future Considerations
The challenge of altering the dismal mortality (and morbidity) of lung cancer, despite novel therapies aimed to “personalize” tumor treatment, continues to be a monumental problem in oncology. In the spirit of entirely novel approaches, harnessing or addressing key components of the host response to malignancy takes us to some promising developments and considerations for future therapeutic design that hopefully will markedly improve our impact on this leading cause of cancer death. Targeting the vascular response to tumor growth now involves co-inhibition of other growth mediators beyond VEGF-A (including PDGF and FGF-2 simultaneously), with inhibition of multiple downstream kinases in attempts to overcome resistance. Biomarkers to identify responders are also under consideration.
While targeted therapies against tumor cells have evolved to respond to the common problem of acquired resistance, along with developments to improve overall survival and quality of life further beyond that offered by combined chemotherapy in metastatic disease, immunotherapy has come online. Challenges in the latter arena in the lung cancer microenvironment include immunosuppression as a result of PD-L1/PD-1 immune checkpoint signaling by tumor–T-cell interactions as well as a suppressive cytokine milieu (TGF-β and IL-10 as examples) within the tumors that promote tolerance by dendritic cells. This inhibits efficient priming of effector CD8+ T cells by tumor antigens. This is in addition to macrophages, MDSCs, and other cells that induce tumor tolerance. Antibodies that target either PD-1 or PD-L1 (or the CTLA-4 molecule that serves a similar T-cell suppressive function) have boosted T-cell responses, with trials in metastatic NSCLC that demonstrate durable curative responses as second- or third-line approaches in 20–25 % of patients. This is exciting and naturally begs the question of how we could target immunity to improve T-cell responses further. Agonist pathways (OX40, 4-1BB, CD27) to promote co-stimulatory T-effector responses (alongside PD-1/PD-L1 axis antagonism) as well as combination approaches are being considered. Novel attempts to target as well as use tumor neo- (unique) antigens are under consideration; and both neo-antigens and self-antigens overexpressed by tumors (e.g., MUC-1 peptide) in vaccines have shown greater efficacy when paired with chemo- or radiation therapy. Finally, a variety of cell-based therapies have been under development to improve specific antitumor T-cell responses. These may include novel exogenous (ex vivo/in vitro) as well as endogenous ways to improve antitumor dendritic cell functions and more direct T-cell therapies using adoptive transfer of cytokine-modified T cells or even engineering of chimeric antigen receptor T (CAR-T) cells against novel lung carcinoma targets. The latter do not depend on presentation of antigen in the context of MHC, which may overcome a significant limitation in NSCLC immunity; however they are limited to single-molecule targets that stand the risk of tumor evolution/heterogeneity and immunologic escape.
References
1.
2.
3.
National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD et al (2011) Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 365(5):395–409CrossRef
4.
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022CrossRefPubMedPubMedCentral
5.
Hall RD, Le TM, Haggstrom DE, Gentzler RD (2015) Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC). Transl Lung Cancer Res 4(5):515–523PubMedPubMedCentral
6.
Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY et al (2015) Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci Rep 5:14273CrossRefPubMedPubMedCentral
7.
Das M, Wakelee H (2014) Angiogenesis and lung cancer: ramucirumab prolongs survival in 2(nd)-line metastatic NSCLC. Transl Lung Cancer Res 3(6):397–399PubMedPubMedCentral
8.
Reck M, Mellemgaard A (2015) Emerging treatments and combinations in the management of NSCLC: clinical potential of nintedanib. Biologics 9:47–56PubMedPubMedCentral
9.
10.
Chang YW, Su CM, Su YH, Ho YS, Lai HH, Chen HA et al (2014) Novel peptides suppress VEGFR-3 activity and antagonize VEGFR-3-mediated oncogenic effects. Oncotarget 5(11):3823–3835CrossRefPubMedPubMedCentral
11.
Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ (2012) Lung cancer in never smokers–a review. Eur J Cancer 48(9):1299–1311CrossRefPubMed
Related posts:
 Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
 Minor-Driver Mutant
Minor-Driver Mutant
 Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
 EGFR Mutant
EGFR Mutant
 PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


