The IGF axis is a tightly controlled endocrine system that regulates cell growth and development, known to have an important function in cancer biology. IGF1 and IGF2 can promote cancer growth in a GH-independent manner both through paracrine and autocrine secretion and can also confer resistance to chemotherapy and radiation. Many alterations of this system have been found in neoplasias, including increased expression of ligands and receptors, loss of heterozygosity of the IGF2 locus and increased IGF1R gene copy number. The IGF1 network is an attractive candidate for targeted therapy, including receptor blockade with monoclonal antibodies and small molecule inhibitors of receptor downstream signaling. This article reviews the role of the IGF axis in the initiation and progression of cancer, and describes the recent advances in IGF inhibition as a therapeutic tool.
- •
The insulin growth factor (IGF) axis is a tightly controlled endocrine system that regulates cell growth and development.
- •
Alterations of this system have been found in neoplasias, including increased expression of ligands and receptors, loss of heterozygosity of the IGF2 locus, and increased IGF1 receptor (IGF1R) gene copy number.
- •
The IGF1 network is an attractive candidate for targeted therapy, including receptor blockade with monoclonal antibodies and small molecule inhibitors of receptor downstream signaling.
- •
Determining the most effective ways to combine IGF1R-targeted therapy with either cytotoxic chemotherapy or other targeted agents as well as identification of predictive biomarkers for patient selection are key future challenges.
Insulin growth factors (IGFs) were initially described as humoral mediators of growth hormone (GH) action. This signaling network is involved in regulation of human development, energy balance, and cell growth. In addition to its well-characterized role in glucose, protein, and lipid metabolism, the complex nature of this hormonal axis continues to attract the attention of scientists and physicians, given the growing body of evidence that supports its critical role in cancer biology. Unlike other trophic factors involved in neoplastic development, IGFs exert endocrine, paracrine, and autocrine effects. The components of this cellular network are summarized in Table 1 .
| IGF1 | Binds IGF1R and hybrid receptors |
| IGF2 | Binds IGF1R, IGF2R, IR-A, and hybrid receptors |
| Insulin | Binds IR-A, IR-B, and IGF1R |
| IGF1R | Binds IGF1 and IGF2 with high affinity and insulin with low affinity |
| IGF2/mannose-6-phosphate receptor | Binds IGF2, lacks intracellular signaling |
| IR-A | Lacks exon 11 (splice variant), binds insulin and IGF2 with high affinity |
| IR-B | Contains exon 11 (splice variant), binds insulin with high affinity |
| IGF1R/IR-A | Hybrid receptor |
| IGF1R/IR-B | Hybrid receptor |
| IGFBP 1–6 (IGFBP1-6) | Binds IGF1 and IGF2, forming tertiary complexes with IGFALS and prolonging the half-life of growth factors |
| IGFBP 7–8 (IGFBP7-8) | Binds IGF1 with low affinity |
| IGFALS | 85-kDa protein, member of leucine-rich repeats superfamily; prolongs half-life of IGFBP-3/5 and IGF1 binary complexes |
In humans, both the insulin and the IGF receptors are composed of 2 globular extracellular alpha subunits that are each linked to a beta subunit and to each other by disulfide bonds. The tyrosine kinase activity resides in the beta subunit. There are also hybrid receptors composed of half an insulin receptor and half an IGF receptor (IRαβ linked to IGF1Rαβ) ( Fig. 1 ). Binding of IGF2 to the IGF2/mannose-6-phosphate receptor (IGF2/M6P-R) triggers internalization and degradation of the ligand, without eliciting downstream signaling. Thus, the IGF2/M6P-R is considered a tumor suppressor because it functions as a scavenger that reduces IGF2 availability to bind IGF1 receptor (IGF1R).
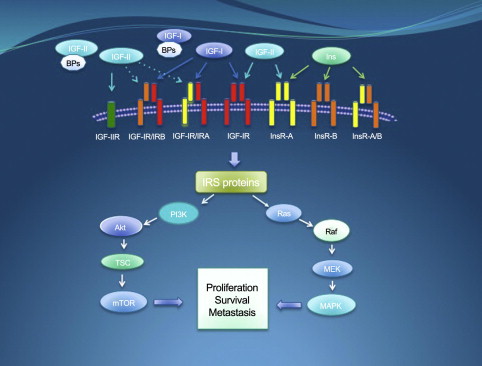
Binding of IGF1 to the alpha subunit of IGF1R triggers a conformational change that causes activation of the catalytic domain in the intracellular beta subunit causing tyrosine autophosphorylation and transphosphorylation (Y1131, Y1135, and Y1136) that enhances its tyrosine kinase activity. These events lead to recruitment of adaptor proteins such as IRS, CRK, and SHC. Downstream signaling is mostly channeled through the MAPK/Ras-Raf-Erk pathway, the phosphatidylinositol-3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway, and the Jak/STAT pathway. Ultimately, activation of IGF1R results in increased cell proliferation and decreased apoptosis.
The central regulation of the IGF system resides in the hypothalamus. Various stimuli lead to growth hormone–releasing hormone or somatostatin secretion. These peptides regulate the secretion of GH by the anterior pituitary gland. Binding of GH to its receptor in the liver results in stimulation of IGF1 synthesis and secretion. In addition, insulin can indirectly increase IGF1 production in part by upregulating GH receptors. Cancer cells, however, can secrete significant quantities of IGF1 in a paracrine or autocrine manner, independently of GH regulation. Thus, the IGF1 axis signals in a hormone-dependent manner to regulate physiologic growth and in a hormone-independent manner in the context of neoplasia.
Individuals with supraphysiologic levels of GH (acromegaly) seem to have an increased risk for cancer development, possibly via an IGF1-mediated mechanism. Conversely, patients with Laron syndrome (IGF1 deficiency and insensitivity to GH) seem to have a lower cancer risk than their relatives.
The influence of circulating IGF1 has been documented in mouse models of tumor development. However, there is a clear distinction between murine and human IGF axis. Although mice present high IGF1 and almost undetectable IGF2 levels postnatally, postnatal human IGF1 level remains at low concentrations. In addition, human IGF2 is not only highly secreted but also functionally relevant beyond birth.
Although IGF2 is also secreted in the liver, this process is not regulated by GH. The IGF2 gene is imprinted. In most tissues, only the paternal allele is expressed. When imprinting of the maternal allele is lost, IGF2 expression is increased. In the case of Beckwith-Wiedemann syndrome, loss of imprinting results in marked IGF2 overexpression resulting in fetal and neonatal overgrowth. Many tumor types harbor biallelic expression of IGF2 via this mechanism, including Wilms tumor, esophageal carcinoma, Ewing sarcoma, rhabdomyosarcoma, colorectal carcinoma, osteosarcoma, prostate adenocarcinoma, hepatocellular carcinoma, and adrenocortical carcinoma.
The bioavailability of IGF1 and IGF2 is highly influenced by their binding to IGF binding proteins (IGFBPs) and the IGF acid-labile subunit (IGFALS). Several factors can increase IGFBP synthesis, including estrogens, retinoids, and vitamin D. In addition, antiproliferative pathways such as transforming growth factor β and p53 also increase IGBP concentration. However, there is also some evidence that shows certain IGFBPs could enhance delivery of these growth factors to the tumor microenvironment. Hyperinsulinism indirectly increases hepatic IGF1 secretion and increases its availability by decreasing IGFBP levels.
Oncogenic transcription factors can also regulate IGFBP expression. EWS-FLI1, an aberrant fusion protein responsible for malignant transformation in Ewing sarcoma family of tumors, binds the IGFBP-3 promoter and decreases its expression. Another example is WT1, a tumor suppressor gene that plays a significant role in the development of Wilms tumor, a pediatric kidney neoplasm. WT1 represses IGF1R expression, thus regulating the activity of the IGF pathway in Wilms tumor.
IGF axis in cancer
Epidemiologic evidence suggests that obesity and type 2 diabetes, both clinical conditions in which hyperinsulinism is present, are associated with increased cancer risk. At least for some tumor subtypes, the same holds true if elevated IGF1 level is detected. Correlation between IGF1R expression and worse outcome has been suggested in different tumor contexts, including colorectal carcinoma, breast cancer, and melanoma.
IGF1R is highly expressed and biologically active in many neoplastic processes. However, gene amplifications were found relatively recently. Increased IGF1R copy number was found in small cell lung cancer. In addition, a high level gain of IGF1R at 15q26 was recently described in pediatric high-grade gliomas, a disease in which IGF2 signaling has a growth-promoting role. Recent studies also suggest elevated insulin receptor A (IR-A) expression in cancer cells.
IGF1R is often necessary for oncogene-mediated malignant transformation. In Ewing sarcoma, IGF1R activity is necessary for transformation of fibroblasts with the EWS/FLI1 fusion transcript, as described in 1997. More recently, silencing of the fusion protein revealed that several microRNAs that negatively regulate expression of proteins in the IGF axis are silenced by EWS/FLI1.
A remarkable aspect of IGF1R biology is its association with resistance to cytotoxic chemotherapy, radiation therapy, and targeted agents. Resistance to cytotoxic agents is documented across tumor types. One of the mechanisms by which this occurs is decreased intracellular concentration of the agent via activation of adenosine triphosphate–dependent efflux pumps such as P-glycoprotein, encoded by the multidrug resistance (MDR) gene. In patients with gastric carcinoma, IGF1R was found to correlate with overexpression of MDR-associated protein 1 and with poor clinical prognosis.
Many preclinical studies have shown sensitization of cancer cell lines to chemotherapy agents by silencing or blocking IGF1R. Although there is some suggestion about AKT activation playing a role in this process, many of the mechanisms involved are still under investigation. In breast cancer cells, IGF1R blockade improved the response to paclitaxel (Taxol) and doxorubicin. Similar results were obtained with non–small cell lung cancer (NSCLC) cells treated with etoposide and carboplatin, atypical teratoid rhabdoid tumor cells treated with doxorubicin and cisplatin, and gastric cancer cells treated with 5-fluorouracil. Other examples include osteosarcoma, colorectal carcinoma, mesothelioma, and esophageal carcinoma. A recent study showed increased sensitivity of glioblastoma cell lines to carmustine and cisplatin when the tumor suppressor gene WT1 was silenced. As described earlier, WT1 negatively regulates IGF1R expression. In hepatocellular carcinoma, IGF1 reduces the cell sensitivity to doxorubicin by changing the redox potential via upregulation of glutathione transferase expression. Silencing of the IGF1R gene enhances sensitivity to DNA-damaging agents such as mitoxantrone, etoposide, and nitrogen mustard in prostate cancer cells. An attractive hypothesis to explain this observation considers IGF signaling as a modulator of XRCC5, a DNA double-strand break repair protein involved in nonhomologous end-joining recombination.
Given its link to DNA repair pathway, it is not surprising that IGF1R inhibition can enhance the effect of radiation therapy. IGF1R knockdown results in decreased ATM expression, whereas defective ATM kinase activity is associated with reduced expression of IGF1R. Both conditions lead to increased radiosensitivity. These findings are consistent with clinical data in breast cancer in which a higher rate of tumor recurrence after surgery and radiation was seen in patients whose primary tumors showed high IGF1R expression.
There are examples of cooperation between hormonal therapy and IGF1 axis inhibition. Antiestrogens, such as tamoxifen, are widely used for the management of estrogen receptor–positive breast cancer. However, 40% of patients do not respond to this treatment. Several factors play a role in tamoxifen resistance. IGF1R is known to cross talk with the estrogen receptor pathway and could be responsible for the resistant phenotype. Tamoxifen-resistant breast cancer cell lines have been shown to have an increased expression of IGF2, which is able to trigger growth and survival signaling via IGF1R. Moreover, IGF1R inhibitors have been able to inhibit growth of tamoxifen-resistant breast cancer cell lines in vitro.
IGF1R inhibition can be combined with other molecularly targeted agents. Some cancer types coexpress IGF1R and epidermal growth factor receptor (EGFR). EGFR family members mediate proliferation, differentiation, and survival in malignant cells. About 40% to 80% of NSCLCs have EGFR overexpression, and 30% of breast cancers overexpress human epidermal growth factor receptor 2 (HER-2). EGFR inhibitors such as erlotinib and gefitinib have been successfully developed, but resistance to therapy often follows initial response. EGFR/IGFR heterodimers that activate the IGF1R signaling pathway have been found after treating NSCLC cell lines with gefitinib. In addition, IGF1R silencing markedly increased apoptosis of gefitinib-treated cell lines. IGF1R has also been found to be a factor in breast cancer resistance to trastuzumab, and there is evidence to suggest that HER-2 phosphorylation is influenced by IGF1R signaling.
Targeting IGF1R
GH Antagonists
Pegvisomant is a genetically engineered GH receptor antagonist used in the treatment of acromegaly. Although there is preclinical evidence of some antitumor activity, its clinical use as an antineoplastic agent has been limited. Somatostatin, the physiologic antagonist of GH, has also been proposed as an anticancer agent in the past.
Ligand Antagonists
IGFPB3 naturally binds the ligands of the IGF axis and decreases their bioavailability in the circulation. Recombinant IGFBP-3 has been proposed as a way to decrease IGF1R signaling, and it showed activity in preclinical models.
MEDI-573 is a human neutralizing IGF1/IGF2 monoclonal antibody that inhibits binding of the growth factors to IGF1R and IR-A. It seems to inhibit IGF1R signaling with virtually no effect in insulin activation of IR-A. Preclinical data show inhibition of tumor growth in vivo using xenografts of high-expressing IGF1R/IR-A cells.
Receptor Antagonists
Several neutralizing antibodies against the IGF1R receptor have been extensively studied, and they continue to be evaluated in many clinical trials. A list of the different currently available agents is shown in Table 2 . There was a significant concern regarding hyperglycemia, because blockade of IGF1R causes a compensatory increase in the levels of GH, which can induce insulin resistance and stimulation of gluconeogenesis. However, hyperglycemia has not been found to be a significant problem in clinical trials using IGF1R blocking antibodies. Available antibodies are either of IgG1 or IgG2 isotype. Isotype differences in terms of side effects, given the different capacity to bind Fc gamma receptors, have not been clearly established yet.
| Agent | Ongoing Trials a | Toxicities | ||
|---|---|---|---|---|
| AMG 479 (Amgen, Thousand Oaks, CA, USA) | Ganitumab | Fully human monoclonal IgG1 | EWS, DSRCT, ovarian carcinoma, CRC, NSCLC; pancreatic carcinoma (phase III) | Thrombocytopenia, hyperglycemia, neutralizing antibodies |
| RG1507 (Roche, Basel, Switzerland) | — | Fully human monoclonal IgG1 | Development discontinued | Hyperglycemia, lymphopenia, CVA |
| IMC-A12 (ImClone, New York, NY, USA) | Cixutumumab | Fully human monoclonal IgG1 | ACC, thymic carcinoma, SCLC, soft tissue sarcomas, osteosarcoma, EWS, HCC, breast cancer, head and neck carcinoma, prostate cancer, hepatocellular carcinoma, islet cell cancer, pancreatic cancer | Hyperglycemia, anemia, infusion reaction |
| MK-0646 (Merck, Whitehouse Station, NJ, USA) | Dalotuzumab | Humanized mouse monoclonal IgG1 | NSCLC, SCLC, CRC, pancreatic carcinoma, breast cancer, neuroendocrine tumors | Thrombocytopenia, GI bleeding, pneumonitis, increased transaminase levels |
| CP-751871 (Pfizer, New York, NY, USA) | Figitumumab | Fully human monoclonal IgG2 | CRC, NSCLC, SCLC, breast cancer; phase III (lung cancer) terminated due to lack of benefit | Hyperglycemia, anemia, cholestasis, hyperuricemia |
| SCH717454 (Schering-Plough, Kenilworth, NJ, USA) | Robatumumab | Fully human monoclonal IgG1 | CRC, EWS, osteosarcoma | — |
| AVE1642 (Sanofi-Aventis, Paris, France) | — | Humanized mouse monoclonal IgG1 | Breast cancer, multiple myeloma, hepatocellular carcinoma | Hyperglycemia, hypersensitivity |
The IGF1 axis has clear biological implications in Ewing sarcoma, and it is not surprising that promising responses have been documented in this group of patients. Durable responses have been achieved in patients with this disease treated with RG1507. In a phase I trial of RG1507 in patients with advanced solid tumors, the drug was well tolerated. Two patients with Ewing sarcoma had confirmed partial responses, and in 13 patients (2 of them with Ewing sarcoma) the disease became stable. In a recent multicenter phase II study of RG1507 in 115 patients with refractory Ewing sarcoma family of tumors, the overall response rate was 10% (1 complete response and 10 partial responses), with a median duration of 29 months. In addition, 8 patients had unconfirmed partial responses. Although the response was overall modest, it is possible that it reflects the need to find an accurate predictive biomarker to determine the patients who are likely to respond to IGF1R blockade. A phase I trial with a different IGF1R blocking antibody, AMG 479, documented a confirmed complete response and a partial response in 2 patients with Ewing sarcoma. Similar results were achieved in a phase I trial of figitumumab in patients with sarcomas.
Another tumor in which blocking IGF1R might be a reasonable strategy is gastrointestinal stromal tumor (GIST). Both wild type and KIT-mutant GIST present high expression of IGF1R, and preclinical activity against these tumors has been promising. Clinical trials to further evaluate this hypothesis are in progress.
A randomized phase III trial of figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with NSCLC was permanently suspended at 681 patients because the futility analysis favored the paclitaxel-carboplatin arm, despite promising previous phase II trials. The study showed that figitumumab does not improve overall survival and increases the risk for severe toxicities, even for patients with nonadenocarcinoma histology. However, patients with circulating levels of IGF1 of greater than 1 ng/mL showed improved overall survival, which again stresses the need of appropriate predictive pharmacodynamic biomarkers in targeted therapy trials.
As mentioned earlier, IGF1R plays a role in resistance to chemotherapy and radiation therapy. Taking advantage of IGF1R inhibition while treating patients with cytotoxic agents and/or radiation therapy is being actively explored. In breast cancer models, there is evidence to support combination of IGF1R inhibitors and EGFR inhibitors, given the cross talk that these 2 relevant pathways have. Moreover, adding downstream signaling inhibition to the receptor blockade to potentiate antitumor effect is a strategy that is being used in several trials in solid tumors. mTOR inhibitors, such as rapamycin, are an interesting choice, because the PI3K/AKT/mTOR pathway is regulated by receptor tyrosine kinases such as IGF1R. In addition, mTOR inhibition induces AKT activation via an IGF-dependent mechanism, supporting the rationale for this combination. Initial results are encouraging, and several trials are under development.
Small Molecule Inhibitors
Tyrosine kinase inhibitors targeting IGF1R have been recently developed. A list of these small molecule inhibitors is depicted in Table 3 . Most of them target insulin receptors in addition to IGF1R. Even though this could bring opportunity for occurrence of metabolic side effects, it is also a chance to improve therapeutic efficacy, given the cross talk between IGF1R and insulin receptors. With one notable exception, these drugs are in early phases of development. Linsitinib efficacy in adrenocortical carcinoma, a disease with high dependence on IGF signaling, is being evaluated in a phase III trial. These drugs can become an attractive therapeutic tool, given a potentially tolerable toxicity profile, the possibility of oral administration, and the theoretical advantage of multitarget inhibition. However, much has to be learned about these agents and the tumor context in which their efficacy can be maximized.
| Agent | Specificity | Ongoing Trials a | Toxicities | |
|---|---|---|---|---|
| XL228 (Exelixis, South San Francisco, CA, USA) | — | IGF1R, SFK, BCR-ABL, FGFR1-3, AURK A, AURK B | Phase I (advanced malignancies/CML) | Hyperglycemia, anorexia, fatigue |
| OSI906 (Linsitinib) (Astellas, Deerfield, IL, USA) | Reversible ATP-competitive inhibitor | IGF1R/IR | NSCLC, ovarian cancer, breast cancer, head and neck carcinoma, HCC, SCLC (phase II), ACC (phase III) | Increased GH and IGF1R concentrations, hyperinsulinemia, hyperglycemia, QTc prolongation, nausea |
| BIIB022 (Biogen, Cambridge, MA, USA) | — | — | Phase I, phase Ib (HCC, NSCLC) | Headache, fatigue, nausea, QTc prolongation, hypertension, GI bleed, glucose level fluctuation |
| AXL 1717 (picropodophyllin) (Axelar, Stockholm, Sweden) | Non-ATP-competitive autophosphorylation inhibitor at Y1136 | High IGF1R selectivity (no IR inhibition) | Phase I | — |
| BMS-754807 (Bristol-Myers, New York, NY, USA) | Reversible, ATP-competitive inhibitor | IGF1R/IR, MET, RON, TrkA, TrkB, AURK A, AURK B | Phase I/II (solid tumors), breast cancer (phase II) | Hypoglycemia, hyperglycemia, fatigue |
| INSM18 (nordihydroguaiaretic acid) (Insmed, Monmouth Junction, NJ, USA) | — | IGF1R/HER-2, 5α-reductase, 15-lipoxygenase, PDGFR | Phase II: prostate cancer | — |
Metformin
Although metformin is not a new targeted agent, its mechanism of action is closely related to the IGF axis. Initially used as an antidiabetic drug that reduces both glucose and insulin plasma concentrations, the role of this agent in cancer therapy is an exciting area of current research that is beyond the limits of this review and has been described elsewhere. Patients treated with metformin seem to have a lower incidence of cancer than untreated controls in epidemiologic studies. This biguanide affects tumor growth by reducing insulin secretion and by inhibiting AMPK-LKB1. Modulation of the IGF axis with a combination of drugs including metformin is promising and is currently under intense investigation.
Targeting IGF1R
GH Antagonists
Pegvisomant is a genetically engineered GH receptor antagonist used in the treatment of acromegaly. Although there is preclinical evidence of some antitumor activity, its clinical use as an antineoplastic agent has been limited. Somatostatin, the physiologic antagonist of GH, has also been proposed as an anticancer agent in the past.
Ligand Antagonists
IGFPB3 naturally binds the ligands of the IGF axis and decreases their bioavailability in the circulation. Recombinant IGFBP-3 has been proposed as a way to decrease IGF1R signaling, and it showed activity in preclinical models.
MEDI-573 is a human neutralizing IGF1/IGF2 monoclonal antibody that inhibits binding of the growth factors to IGF1R and IR-A. It seems to inhibit IGF1R signaling with virtually no effect in insulin activation of IR-A. Preclinical data show inhibition of tumor growth in vivo using xenografts of high-expressing IGF1R/IR-A cells.
Receptor Antagonists
Several neutralizing antibodies against the IGF1R receptor have been extensively studied, and they continue to be evaluated in many clinical trials. A list of the different currently available agents is shown in Table 2 . There was a significant concern regarding hyperglycemia, because blockade of IGF1R causes a compensatory increase in the levels of GH, which can induce insulin resistance and stimulation of gluconeogenesis. However, hyperglycemia has not been found to be a significant problem in clinical trials using IGF1R blocking antibodies. Available antibodies are either of IgG1 or IgG2 isotype. Isotype differences in terms of side effects, given the different capacity to bind Fc gamma receptors, have not been clearly established yet.
Related posts:
 Topoisomerase 1 Inhibitors and Cancer Therapy
Topoisomerase 1 Inhibitors and Cancer Therapy
 Targeting Angiogenesis in Gynecologic Cancers
Targeting Angiogenesis in Gynecologic Cancers
 Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
 mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
 Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
 The Antifolates
The Antifolates
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


