The Hedgehog (Hh) pathway is a signaling cascade that is evolutionally highly conserved and plays an important role in embryonic pattern formation and stem cell response to tissue damage. Given the pivotal role the Hh pathway plays in embryonic development in terms of proliferation and differentiation, it is not surprising that it has also been implicated in tumorigenesis and tumor growth acceleration in a vast variety of malignancies. This article summarizes the mechanism of Hh pathway signal transduction, discusses the models of pathway activation, reviews the clinical data using Hh inhibitors, and discusses challenges to the development of pathway inhibitors.
The Hedgehog (Hh) pathway is a signaling cascade that is evolutionally highly conserved and plays an important role in embryonic pattern formation and stem cell response to tissue damage. The Hh gene was originally identified in 1980 by Christiane Nusslein-Volhard and Eric F. Wieschaus, developmental biologists studying the segmentation pattern of Drosophila melanogaster (fruit fly) embryos. Their genetic analysis found that loss of the gene encoding the secreted protein that is a key regulator of the pathway in fruit flies gives rise to a mutant embryonic phenotype with spiky projections similar to the appearance of a hedgehog. Three Hh homolog genes have since been identified in vertebrates and are termed Sonic hedgehog (Shh) after the popular video game character, and Indian hedgehog (Ihh) and Desert hedgehog (Dhh), both after existing species of hedgehogs. The Hh family of proteins serve a variety of functions in vertebrate embryogenesis, including body patterning, cell migration, growth, differentiation, and survival. This signaling is context dependent, varying based on the receiving cell type, can be short range and long range, and is concentration dependent, typical of a morphogen. The fundamental regulatory role Hh plays in development is underscored by the consequences of pathway misregulation or inhibition, which have been shown to cause severe birth defects, including holoprosencephaly, cyclopia, and microcephaly. In adults, the pathway is much less active and widespread. It is mainly involved in tissue maintenance and repair, including the skin, bone, and intestine.
- •
The Hh pathway is a vital signaling pathway in embryogenesis and more recently has been implicated in the pathogenesis of a vast variety of malignancies.
- •
The key components of the pathway include the Hh ligands Sonic, Indian, and Desert, which bind the receptor PTCH, thereby relieving its inhibition of the receptor SMO, leading to downstream signaling via the GLI transcription factors.
- •
There are multiple models for aberrant Hh pathway activation, including both ligand dependent and independent, as well as a role in tumor stem cell maintenance.
- •
Hh pathway inhibition is an active area of research, with strong preclinical rationale and proof of concept in malignancies, such as basal cell carcinoma, which have constitutive activation of the Hh pathway.
- •
Novel pathway inhibitors have been or are currently being developed to antagonize the Hh pathway and provide a molecularly targeted approach to cancer treatment.
Given the pivotal role the Hh pathway plays in embryonic development in terms of proliferation and differentiation, it is not surprising that it has also been implicated in tumorigenesis and tumor growth acceleration in a vast variety of malignancies. The initial connection between aberrant Hh pathway signaling and cancer was made in a rare condition called Gorlin syndrome, in which patients develop numerous basal cell carcinomas (BCCs) during their lifetimes and are predisposed to other types of malignancies, including medulloblastoma and rhabdomyosarcoma. These patients were noted to have mutations in components of the Hh pathway, leading to its constitutive activation. Other types of cancers, including for example breast, prostate, pancreas, colon, lung, and chronic myelogenous leukemia (CML), have subsequently been shown to have inappropriate Hh pathway activation. Given that misregulation of the Hh pathway has been implicated in such a wide array of disparate malignancies, targeting its signaling has been an active area of research over the past 2 decades. In this article, I summarize the mechanism of Hh pathway signal transduction, discuss the models of pathway activation, review the clinical data using Hh inhibitors, and discuss challenges to the development of pathway inhibitors.
Pathway signal transduction
Signaling within the Hh pathway begins with the secretion of 1 of the 3 Hh proteins: Sonic, Indian, or Desert. The Hh protein undergoes extensive processing, with many modifications and is an unusual secreted protein in that it harbors 2 covalently linked lipid adducts. The precursor protein undergoes initial autoprocessing to release its N-terminal fragment. This peptide is subsequently modified by covalent binding of a cholesterol molecule on its C terminus, conferring a high affinity for the plasma membrane and precluding its further release and spread. The N-terminus then undergoes palmitoylation mediated by an acyl transferase encoded by HHAT. Thus, the ultimate secreted morphogen is termed Hh-Np (“p” standing for processed). The subsequent secretion of the processed protein involves several molecules for its movement, extracellular transport, and release—the best characterized being the large multipass transmembrane protein Dispatched (DISP). It appears that DISP is essential for Hh signaling, as animals with mutations in DISP have accumulation of Hh in producing cells and therefore downstream signaling of the pathway is lost.
Once secreted from the producing cell, Hh binds on the receiving cell to the Patched protein (PTCH), a 12-transmembrane protein, and initiates the signaling cascade. The simplest model is that Hh binds PTCH, alleviating PTCH inhibition of the 7-transmembrane G-protein-coupled receptor–like protein Smoothened (SMO), thereby enabling signal transduction to the nucleus ( Fig. 1 ). Conversely, in the absence of ligand binding, PTCH catalytically inhibits the activity of SMO; however, the exact mechanism of the interaction is unclear. It is known from tissue-cultured cells that PTCH inhibits SMO at substoichiometric concentrations. One proposed mechanism for PTCH inhibition of SMO may involve the transport by PTCH of a small molecule regulator of SMO movement or activity. Candidates for this small molecule include PI4P, lipoproteins, and provitamin D3. Another possible mechanism is that PTCH represses SMO by affecting its localization to the cell surface. Specifically, it is thought that PTCH prevents SMO translocation into the primary cilium, a microtubule-based antennalike structure that emanates from the surface of virtually all cells in the mammalian body. The primary cilium is a sensory organelle that receives both mechanical and chemical signals from other cells and the environment, and transmits these signals to the nucleus to elicit a cellular response. It has been shown to play a pivotal role in the Hh transduction pathway. For example, mutations in intraflagellar transport, which normally functions in retrograde and anterograde movement of molecules within the primary cilium, results in mice with Hh loss-of-function phenotypes. When exposed to Hh ligand, PTCH, which is normally present in the cilia, becomes internalized, thereby allowing SMO to move from internal vesicles all the way up along the shaft of the cilium.
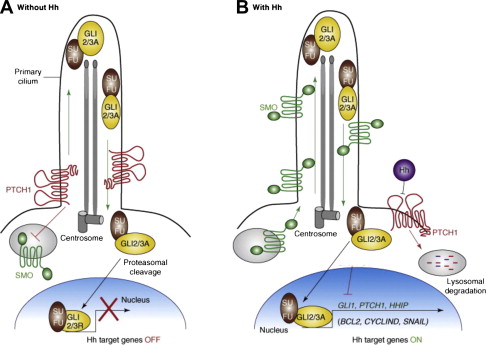
Once Hh binds to PTCH and thereby alleviates repression of SMO, a signaling cascade is initiated through activation of the GLI family of zinc transcription factors, thereby translating the extracellular stimulus into defined transcriptional programs in a context-dependent and cell-type specific manner. Unlike Drosophila , which have a single GLI protein that is encoded by the cubitus interruptus gene, vertebrates have 3 GLI transcriptions factors (GLI 1–3). GLI2 and 3 are bifunctional transcription factors, functioning as either activators or repressors depending on their modification, processing, and nuclear trafficking. Because GLI1 lacks the N-terminal repressor domain, it functions exclusively as an activator. It is also most highly dependent on active Hh signaling and therefore is the most often used surrogate for pathway activation. GLI family members are phosphorylated by protein kinase A (PKA) and glycogen synthase 3 (GSK3) in the absence of Hh signaling, but are relieved from PKA/GSK3-mediated phosphorylation in the presence of Hh signaling. In the absence of Hh activation, GLI2 is degraded, owing to phosphorylation-mediated ubiquitylation, and GLI3 is processed into a repressor. Once the Hh pathway is activated, GLI family members are stabilized, translocated into the nucleus, and bind the consensus binding site (5′-TGGGTGGTC-3′) in the target gene promoter. Hh signaling seems to be dependent on the relative balance of GLI activator and repressor forms. The activity of GLI transcription factors is regulated at several different levels, including nuclear-cytoplasmic shuttling, ubiquitination, protein degradation, and the transcriptional activity of GLI molecules. One well-characterized negative regulator of Hh signaling is the Suppressor of Fused (SUFU) protein, which acts as a tumor suppressor gene. It associates with and inhibits GLI molecule function and is required for GLI3 processing. The Hh target genes are numerous and involved in a wide variety of cellular functions, including control of cell proliferation, survival, epithelial-to-mesenchymal transition (EMT), maintenance of stemness, and cell fate determination. In addition, GLI, PTCH, and Hh-interacting protein are not only components of the pathway, but are also target genes themselves and involved in several regulatory feedback loops.
Linking the H h pathway and human cancer
Similar to its role in Drosophila , the Hh pathway in humans was initially implicated in embryogenesis and development as an essential signaling pathway. Unlike in the embryo or neonate, Hh activation was found to be much more limited in the adult and restricted to areas of tissue damage and repair. More recently, however, aberrant Hh pathway signaling has been implicated in cancer formation and growth. The link was first made in 1996 when mutations in PTCH were discovered to be associated with a rare hereditary form of BCC, called basal cell nevus syndrome or Gorlin syndrome. Gorlin syndrome is an autosomal dominant genetic disease in which patients develop numerous BCCs during their lifetime and are at an increased risk of other tumors, including medulloblastoma, a tumor of the cerebellar progenitor cells, and rhabdomyosarcoma, a muscle tumor. These patients have a germline mutation in 1 allele of the PTCH gene, and basal-cell tumors from these patients lack the remaining normal PTCH gene. Without functional PTCH, SMO is freed from repression and the Hh pathway becomes constitutively active. The association between the Hh pathway and tumorigenesis was further supported by the identification of aberrant activation in sporadic BCCs. Most BCC tumors have either inactivating mutations in PTCH1 (>85%) or, less commonly, activating mutations in SMO. Since the initial connection between the Hh pathway and carcinogenesis in BCCs, aberrant signaling and/or reactivation of the cascade have been noted in a wide and disparate number of cancers, including both solid tumor and hematologic malignancies.
Linking the H h pathway and human cancer
Similar to its role in Drosophila , the Hh pathway in humans was initially implicated in embryogenesis and development as an essential signaling pathway. Unlike in the embryo or neonate, Hh activation was found to be much more limited in the adult and restricted to areas of tissue damage and repair. More recently, however, aberrant Hh pathway signaling has been implicated in cancer formation and growth. The link was first made in 1996 when mutations in PTCH were discovered to be associated with a rare hereditary form of BCC, called basal cell nevus syndrome or Gorlin syndrome. Gorlin syndrome is an autosomal dominant genetic disease in which patients develop numerous BCCs during their lifetime and are at an increased risk of other tumors, including medulloblastoma, a tumor of the cerebellar progenitor cells, and rhabdomyosarcoma, a muscle tumor. These patients have a germline mutation in 1 allele of the PTCH gene, and basal-cell tumors from these patients lack the remaining normal PTCH gene. Without functional PTCH, SMO is freed from repression and the Hh pathway becomes constitutively active. The association between the Hh pathway and tumorigenesis was further supported by the identification of aberrant activation in sporadic BCCs. Most BCC tumors have either inactivating mutations in PTCH1 (>85%) or, less commonly, activating mutations in SMO. Since the initial connection between the Hh pathway and carcinogenesis in BCCs, aberrant signaling and/or reactivation of the cascade have been noted in a wide and disparate number of cancers, including both solid tumor and hematologic malignancies.
Models of pathway activation in cancer
There are 3 basic models that have been proposed for Hh pathway activity in cancer ( Fig. 2 ). The first elucidated was the Hh ligand-independent cancers, such as BCCs, and were termed type I cancers. These tumors have pathway-activating mutations that lead to constitutive activation of the Hh pathway. Type II cancers are ligand-dependent and autocrine (or juxtacrine), so that Hh is both produced and responded to by the same (or neighboring) tumor cells, whereas Type III cancers are also ligand dependent, but use paracrine signaling. In this model, Hh is produced by the tumor epithelium and received by the surrounding stroma (analogous to epithelial-to-mesenchymal transition in early development), which then feeds back growth and/or survival signals to the tumor. A variation to this model has recently been described, termed “reverse paracrine” signaling or Type IIIb, where Hh is secreted from stromal cells to the receiving cells in a tumor.
In addition to the aforementioned 3 models, there is an alternative model that implicates aberrant Hh pathway activation in cancer stem cells (CSCs). CSCs comprise a small subset of cells in a tumor that are characterized by being able to self-renew and initiate tumor spread. They are typically resistant to chemotherapy, possibly contributing to relapse. Similar to its role in normal stem cell activity, the Hh pathway has been shown to be active in several types of CSCs, including breast, glioma, pancreatic, multiple myeloma, and CML. The exact mechanism of Hh deregulation within CSCs is still unclear and may rely on autocrine, paracrine, and/or ligand-independent signaling. What is clear, however, is that Hh inhibition appears to deplete this tumorigenic population in animal models and therefore holds great potential promise in the clinic.
Discovery of the first H h inhibitor: cyclopamine
The discovery of the first Hh pathway inhibitor is an interesting story and dates back to the 1950s when an increased incidence of congenital malformations, including cyclopia, was noted in newborn lambs. These teratogenic effects were linked to pregnant ewes consuming a corn lily plant ( Veratrum californicum ). Analysis of the plant led to isolation and identification of the principal teratogen, termed cyclopamine, a steroidal alkaloid. Following the discovery of the Hh pathway first in Drosophila and later in humans and the understanding that mutations within this signaling cascade can lead to deformities, such as holoprosencephaly, which can include cyclopic features, Cooper and colleagues demonstrated that cyclopamine acts through inhibition of the Hh pathway. The elucidation of cyclopamine as an Hh pathway inhibitor took decades and has since served as a powerful tool to help understand the role the Hh pathway plays in diseases, including cancer.
Cyclopamine inhibits the Hh pathway by binding to, and inactivating SMO. It has been used extensively in preclinical studies; however, because of its low affinity, poor oral bioavailability, and suboptimal pharmacokinetics, it has not moved forward in the clinic. Furthermore, in addition to inactivating SMO, cyclopamine may also have “off-target” effects, as cells without the SMO receptor have been shown to undergo apoptosis and often very high concentrations of cyclopamine are required to inhibit cell proliferation. Given the multiple impediments to the clinical use of cyclopamine, as well as the potential importance of the Hh pathway in a vast array of pathologies, additional inhibitors with improved pharmacologic properties have since been identified via high-throughput screening of small-molecule libraries. Although the screens were designed to probe the entire Hh signaling cascade, most of the identified inhibitors have been reported to target SMO itself. SMO seems to be the most druggable component of the pathway, whereas other components are more structurally complex and inaccessible.
H h pathway inhibitors
Among the various Hh pathway inhibitors (HPIs), vismodegib (GDC-0449; Curis/Genentech) is the farthest along in development. Vismodegib is a synthetic small-molecule inhibitor of the Hh pathway that binds to and inhibits SMO. It was designed to be more potent and have more favorable pharmaceutical properties than the steroidal alkaloid cyclopamine. Phase I clinical testing is completed and vismodegib is now being evaluated in multiple phase II studies, some of which have recently been reported, including colon, ovarian, and BCCs (see later in this article). Other synthetic SMO inhibitors include BMS-833923 (XL139; Exelexis/Bristol-Myers Squibb), LDE225 and LEQ506 (Novartis), PF-04449913 (Pfizer), and LY2940680 (Eli Lily), which have either completed or are nearing completion of phase I testing. IPI-926 (Infinity) is the only semisynthetic SMO inhibitor currently in clinical testing. It was derived from cyclopamine and demonstrates improved potency and physiochemical properties, including acid stability and aqueous solubility. Phase I single-agent findings have been reported and currently combination phase I and phase II testing is ongoing for chondrosarcoma, and pancreatic and head and neck cancers ( Table 1 ).
| Drug | Tumor Target | Other Agent(s) | Phase | Design | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| GDC-0449 | Pancreas | — | II (single arm) | Preoperative GDC-0449 | NCT01096732 |
| Pancreas | Gemcitabine, nab-paclitaxel | II (single arm) | Metastatic disease treated with GDC-0449 + chemotherapy | NCT01088815 | |
| Pancreas | Gemcitabine | Unspecified | Pilot study to investigate pancreatic stem cells before & after treatment with GDC-0449 + gemcitabine | NCT01195415 | |
| Pancreas | Erlotinib, gemcitabine | I | Metastatic disease treated with GDC-0449 + erlotinib ± gemcitabine | NCT00878163 | |
| Pancreas | Gemcitabine | II (randomized, placebo controlled) | Metastatic disease treated with gemcitabine ± GDC-0449 | NCT01064622 | |
| Small cell lung cancer | Cisplatin (cis), etoposide (E) | II (randomized) | Three arms: cis+E, cis+E+ GDC-0449, cis+E + cixutumumab | NCT00887159 | |
| Gastroesophageal | 5-Fluoruracil, oxaliplatin | II (randomized, placebo controlled) | Advanced gastroesophageal cancer treated with FOLFOX ± GDC-0449 | NCT00982592 | |
| Prostate | Leuprolide acetate or goserelin | Ib/II (randomized) | Preoperative GDC-0449 + androgen ablation compared with androgen ablation alone followed by radical prostatectomy for locally advanced prostate adenocarcinoma | NCT01163084 | |
| Medulloblastoma | — | I | Recurrent or refractory medulloblastoma in pediatric patients | NCT00822458 | |
| Medulloblastoma | — | II (single arm) | Recurrent or refractory medulloblastoma in pediatric patients, stratified by IHC for Hh pathway | NCT01239316 | |
| Medulloblastoma | — | II (single arm) | Recurrent or refractory disease treated with GDC-0449 | NCT00939484 | |
| Sarcoma | RO4929097 | Ib/II | Advanced or metastatic sarcomas treated with combination GDC-0449 + gamma secretase inhibitor | NCT01154452 | |
| Chondrosarcoma | — | II | Metastatic or unresectable chondrosarcoma treated with GDC-0449 | NCT01267955 | |
| Multiple myeloma | — | Ib | High-risk myeloma in first remission following autologous stem cell transplantation | NCT01330173 | |
| Basal Cell Carcinoma (BCC) | — | II | 2-cohort trial evaluating the efficacy & safety of GDC-0449 in operable BCC | NCT01201915 | |
| BCC | — | II (single arm) | Locally advanced or metastatic BCC | NCT01367665 | |
| IPI-926 | Pancreas | Gemcitabine | Ib/II | Metastatic disease patients randomized in phase II to gemcitabine ± IPI-926 | NCT01130142 |
| Pancreas | 5-Fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) | I | Advanced disease treated with FOLFIRINOX + IPI-926 | NCT01383538 | |
| Head and neck | Cetuximab | I | Advanced disease treated with combined cetuximab + IPI-926 | NCT01255800 | |
| Chondrosarcoma | — | II (randomized, placebo controlled) | Unresectable disease treated with IPI-926 or placebo | NCT01310816 | |
| Myelofibrosis | — | II | Single-arm study with primary end point of response rate | NCT01371617 | |
| LDE225 | BCC | — | II | Randomized double-blind study of efficacy and safety of 2 dose levels of LDE225 in unresectable disease | NCT01327053 |
| BCC | — | II (randomized, placebo controlled) | Adult patients with nevoid BCC syndrome | NCT01350115 | |
| Pancreas | Gemcitabine | I/II | Neoadjuvant combination therapy in borderline resectable pancreatic cancer | NCT01431794 | |
| BMS-833923 | CML | Dasatinib | I | Dose-finding study in CML patients with resistance or suboptimal response to a prior TKI | NCT01218477 |
| Gastroesophageal | Cisplatin, capecitabine | Ib | 1st line treatment of advanced disease | NCT00909402 | |
| Small cell lung cancer (SCLC) | Carboplatin, etoposide | Ib | Extensive stage SCLC treated with BMS-833923 + carboplatin + etoposide followed by maintenance BMS-833923 | NCT00927875 | |
| PF-04449913 | CML/hematologic malignancies | Dasatinib or bosutinib | I | PF- 04449913 in select hematologic malignancies or in combination with dasatinib or bosutinib in CML | NCT00953758 |
| Solid tumors | — | I | Advanced solid tumors treated with single agent | NCT01286467 | |
| LY2940680 | Solid tumors | — | I | Advanced solid tumors treated with single agent | NCT01226485 |
| Itraconazole | BCC | — | Pilot | To determine if 3 weeks of oral or topical itraconazole reduces BCC biomarkers | NCT01108094 |
| SCLC | Pemetrexed | Randomized phase II | Previously treated SCLC treated with pemetrexed ± itraconazole | NCT00769600 |
Although there are definite differences among the various SMO inhibitors in terms of structure, they otherwise have similar profiles. They are all oral selective small molecule inhibitors with high potency (half maximal effective concentration <20 nM), long half-lives (24–168 hours), and a very manageable side-effect profile. The most common toxicities include fatigue, dysgeusia, myalgias, alopecia, and nausea, which may be a class effect. They also have all shown to have exposure-dependent target inhibition as measured by GLI expression.
Apart from SMO inhibitors, there are other Hh pathway components that are being targeted for inhibition. One of the more challenging targets has been the Hh pathway ligand Shh, an extracellular protein that is upstream of SMO and undergoes extensive lipid modification, making it a challenge to access. The macrocylic small molecule robotnikinin is the first reported inhibitor of Shh. It was discovered through a small-molecule microarray-based screen of a bacterially expressed biologically active Shh N-terminal fragment (ShhN) and then subsequently optimized for better binding and dissociation time. It binds to ShhN at concentrations between 1.56 and 25.00 μM with a Kd value of 3.1 μM. It was shown to block transcription of Hh pathway targets in primary keratinocytes treated with Shh compared with that seen in mock-treated controls. It did not prevent transcription triggered by small-molecule agonists of SMO, suggesting that the compound acts earlier in the pathway than SMO. Although still in preclinical development, robotnikinin and other small-molecule inhibitors of Shh may ultimately play an important role in the treatment of ligand-dependent tumors. There are multiple other inhibitors that act downstream of SMO on various different facets of the Hh pathway. Inhibition of GLI-mediated transcription is an attractive target, as it is theoretically efficacious irrespective of the mode of Hh pathway activation. Two low-molecular-weight compounds, GANT58 and GANT61, have been identified, which act at the nucleus to block GLI function. Although they are from different chemical classes, they both are potent and selective inhibitors of GLI with a half maximal inhibitory concentration of ∼5 μM. Furthermore, they both block cell growth in an in vivo xenograft prostate cancer model. More recently, 4 additional Hh pathway inhibitors, which act downstream of SMO, have been characterized. Termed HPI-1 through HPI-4, each has a unique mechanism of action, involved in GLI processing, GLI activation, and primary cilia formation, suggesting that multiple steps in GLI regulation are pharmacologically targetable.
Additionally, 2 well-known and widely used agents approved by the Food and Drug Administration (FDA) for separate indications have been shown to inhibit the Hh pathway. Both itraconazole, a commonly used antifungal, and arsenic trioxide, an important treatment for acute promyelocytic leukemia, have demonstrated preclinical evidence for Hh pathway inhibition. Arsenic trioxide’s mechanism of action is thought to be through inhibition of GLI transcriptional effectors, blocking Hh-induced ciliary accumulation of GLI2 and thereby reducing steady-state levels, whereas itraconazole has been shown to act at the level of SMO by a mechanism that is distinct from that of cyclopamine and other known SMO antagonists, and prevents the ciliary accumulation of SMO normally caused by Hh stimulation. There is currently a clinical trial under way with itraconazole in patients with BCCs, building on this new understanding of an established drug (see Table 1 ).
Related posts:
 Topoisomerase 1 Inhibitors and Cancer Therapy
Topoisomerase 1 Inhibitors and Cancer Therapy
 Targeting Angiogenesis in Gynecologic Cancers
Targeting Angiogenesis in Gynecologic Cancers
 Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
 mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
 Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
 The Antifolates
The Antifolates
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


