Fig. 1.1
Chemical structure of rapamycin and rapalogs. Rapalogs have the indicated O-substitutions at the C-40 position of rapamycin (red circle)
Temsirolimus, formerly known as CCI-779; Torcel®, Wyeth Pharmaceuticals, now Pfizer Pharmaceuticals, is a dihydroxymethyl propionic acid ester prodrug of rapamycin. This modification renders temsirolimus more water soluble than rapamycin and thus it can be administered intravenously. Upon injection, temsirolimus is rapidly converted to rapamycin, which is responsible for most, if not all, of its pharmacological effects.
Everolimus, an oral, water-soluble rapalog formerly known as RAD001; Afinitor®; Novartis Pharmaceuticals, has an O-(2-hydroxyethyl) chain substitution at position C-40 and is also converted to rapamycin.
Ridaforolimus, a water-soluble, parenteral formulation formerly known as AP23573; Ariad Pharmaceuticals, has a phosphine oxide substitution at the same position of the lactone ring of rapamycin.
Zotarolimus, the first rapalog developed specifically for local delivery from stents for the prevention of restenosis, has a tetrazole ring in place of the native hydroxyl group at position 42 of rapamycin (Fig. 1.2). The compound, developed by Abbott Laboratories (Chicago, Illinois), is very lipophilic, which is more conducive for local delivery and prevents rapid release into the systemic circulation.
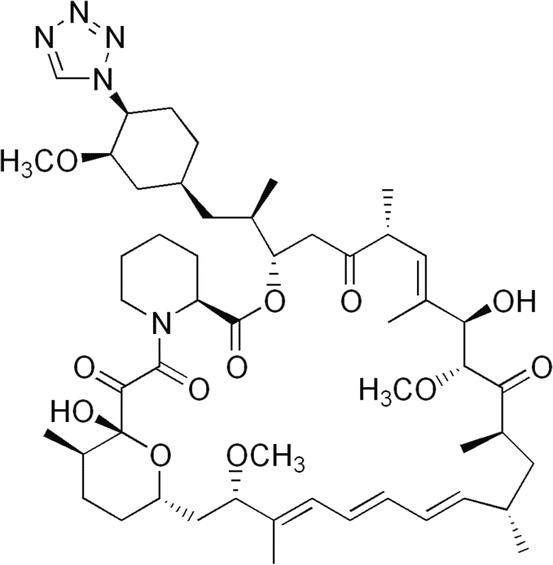
Fig. 1.2
Chemical structure of zotarolimus, a highly lipophilic rapalog developed specifically for local delivery from stents for the prevention of restenosis. Zotarolimus has a tetrazole ring in place of the native hydroxyl group at position 42 of rapamycin
1.7 Prevention of Organ Rejection Following Allogeneic Organ Transplant Rejection
Soon after rapamycin was demonstrated to inhibit mTOR-mediated signal transduction pathways in lymphocytes, it was shown to block post-receptor immune responses to co-stimulatory signal 2 during G0 to G1 transition, as well as cytokine signal 3 during progression through the G1 phase. In addition, rapamycin was demonstrated to inhibit interleukin-2- and interleukin-4-dependent proliferation of T- and B-lymphocytes, resulting in the suppression of new ribosomal protein synthesis [32, 33]. Because of these potent immunosuppressive effects, rapamycin (sirolimus, Rapamune®, Wyeth Pharmaceuticals) was evaluated in the setting of renal transplantation to treat and prevent organ rejection in the 1990s. The promising results of a phase 1 clinical trial of sirolimus by Kahan and colleagues led to the first randomized, placebo-controlled, multicenter phase 2 clinical trial, which evaluated the combination of cyclosporine A and corticosteroids plus either sirolimus or placebo in the setting of acute renal allograft rejection [34, 35]. This study demonstrated that the incidence of biopsy confirmed acute renal allograft rejection within the first 6 months after renal transplantation was significantly reduced in the sirolimus group. Moreover, patients receiving sirolimus plus a reduced dose of cyclosporine A had significantly better renal function, indicating that co-administration of these agents permit cyclosporine A dose reduction without jeopardizing organ function. Encouraged by these results, two large multicenter phase 3 trials confirmed the phase 2 findings ultimately leading to first regulatory approval of sirolimus. In September 1999, the United States Food and Drug Administration (FDA) granted regulatory approval to sirolimus combined with cyclosporine A and corticosteroids for the prevention of organ rejection following renal transplantation [36, 37]. Soon after, sirolimus received regulatory approval by the European Medicines Agency (EMA) in 2000 as an alternative to calcineurin inhibitors for maintenance immunosuppression to prevent renal graft rejection. After further studies indicated that patients receiving sirolimus plus corticosteroids without cyclosporine A had significantly better graft survival and function, the FDA subsequently approved sirolimus without cyclosporine A; however, the combination is recommended in the early post-transplantation setting.
Everolimus was subsequently approved by the EMA in 2003 and FDA in 2010 for use with low-dose cyclosporine, basiliximab, and corticosteroids to prevent organ rejection in adult renal transplant patients who have a low-to-moderate immunologic risk based on the results of a single multicenter randomized phase 3 trial. The study demonstrated that everolimus-based therapy is effective at preventing acute organ rejection while using a 60 % lower dose of cyclosporine A compared with the control regimen (mycophenolic acid, cyclosporine, and corticosteroids) [38].
Both EMA and FDA approved everolimus for the prophylaxis of organ rejection in adult patients receiving a liver transplant in 2012 and 2013, respectively. The approval was based on the results of a phase 3 trial, which showed that everolimus combined with reduced-dose tacrolimus led to comparable efficacy and superior renal function than standard-dose tacrolimus at 12 months post-transplantation [38]. In addition, a large independent registry study of nearly 70,000 patients who received a non-renal solid organ transplant between 1990 and 2000 showed that the incidence of chronic renal failure was greater in liver transplant recipients than in recipients of all other solid organ transplants, except intestinal transplants, thereby supporting the previous pivotal trial results. Since calcineurin inhibitors, such as tacrolimus, are part of the standard-of-care treatment regimen for immunosuppression in liver transplantation and may contribute to impaired renal function, the opportunity to lower calcineurin inhibitor exposure by co-treatment with everolimus was viewed as quite favorable.
The EMA approved everolimus for prophylaxis of organ rejection in adult patients at low-to-moderate immunological risk receiving an allogeneic cardiac transplant in 2003 [39].
1.8 Drug-Eluting Cardiac Stents
After the introduction of balloon angioplasty in 1977, intracoronary arterial stenting was perhaps the most important development in the field of percutaneous coronary arterial revascularization; however, post-angioplasty restenosis, or lumen re-narrowing, several months after the index procedure, became a formidable challenge to the benefits of this intervention, often resulting in recurrent symptoms, repeat intervention, coronary bypass graft surgery, and myocardial infarction [40]. Stent-induced restenosis involves a complex interplay of biological events. We now know that the placement of cardiac arterial stents results in endothelial injury, as well as deeper injury due to lacerations of the arterial wall. Further, such injury is now known to stimulate the accumulation of macrophages around the stent, and smooth muscle cells proliferate and migrate from the underlying vessel wall [41]. Despite the scaffolding effect of the stent, the smooth muscle cells accumulate gradually, impinging on the lumen. To address this issue, developers of drug-eluting cardiac arterial stents used the devices as tools to deliver medications directly to the arterial wall. While initial efforts were unsuccessful, the elution of drugs with certain specific physicochemical properties from the stent was shown in 2001 to achieve high concentrations of the drug locally, directly at the target lesion, with minimal systemic side effects [42]. As currently used in clinical practice, “drug-eluting” stents refer to metal stents that elute a drug designed to limit the growth of neointimal scar tissue, thus reducing the likelihood of stent restenosis [42].
In vivo studies in allograft and angioplasty models in the late 1990s demonstrated the effectiveness of sirolimus in preventing tissue hyperplasia following vascular injury and led to consideration and evaluation for the prevention of restenosis [43, 44]. The First-in-Man feasibility study conducted in Sao Paulo, Brazil, and Rotterdam, the Netherlands, showed the CYPHER® sirolimus-eluting stent (Cordis Corporation, Johnson and Johnson, Warren, NJ) to be remarkably effective in preventing restenosis [45]. These early results were followed by the unprecedented findings from the RAVEL trial, the first double-blind, randomized, controlled phase 3 trial of a drug-eluting stent [46]. These studies resulted in CE Mark approval for the CYPHER® sirolimus-eluting stent in Europe in April 2002 and subsequently in the United States in July 2013. The initial results were soon after replicated in three additional randomized, controlled phase 3 trials – SIRIUS, E-SIRIUS, and C-SIRIUS [47–49]. Since the preliminary results of the First-in-Man feasibility study were presented, the CYPHER® stent has been used to treat several million patients in more than 80 countries.
Several other rapalogs have been evaluated as antiproliferative components in drug-eluting cardiac arterial stents. Abbott Laboratories (Chicago, Illinois) specifically developed the highly lipophilic rapalog zotarolimus (formerly named ABT-578) for use in drug-eluting stents with phosphorylcholine as the carrier. However, their ZoMaxx® stent, a stainless steel and tantalum-based stent in which phosphorylcholine slowly releases zotarolimus, showed less neointimal inhibition, manifesting as poor clinical performance, when compared with paclitaxel-eluting stents in a long-term follow-up of a randomized, controlled phase 3 trial [50]. Zotarolimus was licensed to Medtronic (Minneapolis, Minnesota), which is the basis for their Endeavor® drug-eluting stent whose cobalt alloy structure uses phosphorylcholine as a carrier for zotarolimus. The Endeavor® stent was approved for use in Europe in 2005 and the United States in 2014 [40]. Lastly, Guidant, Corporation (Indianapolis, Indiana) received EMA approval for the XIENCE® stent V coronary stent system that elutes everolimus in 2006; regulatory approval occurred in the United States in 2008. XIENCE® is currently marketed by Abbott Laboratories.
1.9 Malignant Diseases
Much of the scientific foundation for the various regulatory approval discussed in this section will be highlighted in greater detail in specific sections throughout this book.
Temsirolimus (Torisel®, Wyeth Pharmaceuticals) became the first rapalog approved in the United States, Europe, and elsewhere in 2007 to treat with advanced renal cancer based on the results of a multicenter phase 3 trial in the first-line treatment setting in which treatment-naïve patients with advanced disease and poor prognosis were randomized to treatment with either interferon-alpha, temsirolimus, or the combination of both agents [51]. There was a statistically significant longer overall survival for patients treated with temsirolimus than those in the interferon-alpha monotherapy arm, as well as a statistically significant longer progression-free survival time for patients treated with temsirolimus, whereas the combination of both agents resulted in greater toxicity and no statistically significant difference in overall survival when compared with interferon-alpha alone. In 2009, temsirolimus received market authorization in the European Union for treatment of relapsed and refractory mantle cell lymphoma on the basis of a multicenter phase 3 trial comparing two different temsirolimus dosing regimens with an investigator’s choice of therapy in 162 patients with relapsed and/or refractory mantle cell lymphoma [52]. Patients treated with temsirolimus had a statistically significant improvement in the primary endpoint of progression-free survival compared with those in the investigator’s choice arm, and temsirolimus treatment was associated with statistically significant advantages over investigator’s choice in the secondary endpoint of overall response rate. Temsirolimus was not associated with a significantly longer overall survival, a secondary endpoint.
Everolimus has been approved as a single agent in several advanced malignancies in both the United States and Europe. Both FDA and EMA approved everolimus in 2009 for patients with advanced renal cell carcinoma after failure of a vascular growth factor receptor targeted therapy, based on a statistically significant improvement in progression-free survival compared with placebo [53]. Everolimus was subsequently approved by both FDA and EMA in 2011 for the treatment of adults with metastatic or locally advanced progressive neuroendocrine tumors located in the pancreas based on the results of a phase 3 multicenter trial (RADIANT-3) involving 410 patients randomized to treatment with either everolimus or placebo [54]. Progression-free survival, the primary endpoint of the study, was significantly longer in patients receiving everolimus treatment compared with placebo (11 versus 4.6 months). Everolimus treatment was associated with a low rate of adverse events. Lastly, everolimus became the first rapalog to receive regulatory approval as a modulator of hormone sensitivity in combination with a hormonal therapy in 2012. Both FDA and EMA approved everolimus in combination with exemestane to treat certain postmenopausal women with advanced hormone-receptor positive, HER2-negative breast cancer whose disease had recurred or progressed after treatment with letrozole or anastrozole. The safety and effectiveness of everolimus were evaluated in a clinical study of 724 postmenopausal women with advanced estrogen receptor-positive and HER2-negative and had previously received treatment with the aromatase inhibitors letrozole or anastrozole [55]. Patients were randomized to receive treatment with exemestane plus either everolimus or placebo. Patients treated with everolimus plus exemestane had a 4.6 month improvement in progression-free survival compared to patients receiving the placebo plus exemestane.
Clinical evidence of antitumor activity has been noted with various other rapalogs in a wide range of other malignancies including endometrial and ovarian cancers and soft-tissue sarcoma. The largest effort in, as of yet, unapproved indications has been in patients with advanced sarcoma. The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial was a randomized (1:1), placebo-controlled, double-blind phase 3 study of oral ridaforolimus in 771 patients with metastatic soft-tissue or bone sarcomas who previously had a favorable response to chemotherapy [56]. The study achieved its primary endpoint of improving progression-free survival, achieving a statistically significant (28 %) reduction in the risk of progression or death observed in those treated with ridaforolimus compared to placebo. Median PFS was 17.7 weeks for those treated with ridaforolimus compared to 14.6 weeks in the placebo group (hazard ratio, 0.72; p = 0.0001).
1.10 Tuberous Sclerosis Complex
In 2010, everolimus received accelerated approval in the United States for the treatment of patients with subependymal giant cell astrocytoma (SEGA) associated with the TSC, as previously discussed in this chapter, who require therapeutic intervention and are not candidates for curative surgical resection. The approval was based on a single arm trial that demonstrated a 50 % or greater reduction in SEGA tumor volume in 9 (32 %) of 28 children and adults [57]. The EMA followed up with an approval for everolimus for this indication in 2011. The FDA subsequently expanded its approval to children younger than 3 years of age in 2012 based on the results of a randomized double-blind placebo-controlled trial in pediatric and adult patients with SEGA. In this trial, 78 children and adults (median age, 9.5 years [range, 0.8-26]) were randomly assigned to receive treatment with everolimus and 39 to receive placebo. SEGA responses were observed in 27 (35 %) of 78 everolimus-treated patients and none of the 39 patients treated with placebo (p < 0.0001). The median response duration was 5.3 months (range, 2.1–8.4 months) in patients treated with everolimus [58].
Everolimus received approval by both FDA (accelerated) and EMA for the treatment of adults with renal angiomyolipoma associated with TSC who do not require immediate surgery in 2012. The approval was based on durable reductions in tumor volume in everolimus-treated patients in a randomized (2:1), double-blind, placebo-controlled trial conducted in 118 patients with renal angiomyolipoma as a feature of the TSC (n = 113) or sporadic lymphangioleiomyomatosis (n = 5) [59]. Confirmed objective responses in renal angiomyolipoma were noted in 33 (41.8 %) patients treated with everolimus, whereas no patient in the placebo arm responded (p < 0.0001) [59]. The median response duration was 5.3+ months (range, 2.3+ to 19.6+ months).
Based on the association of TSC with mental retardation, autism, seizure disorders, and neuropsychological problems, including long-term and working memory deficits, researchers have developed genetically engineered mice with a heterozygous inactivating mutation in the TSC2 gene (Tsc2+/− mice) that confer deficits in learning and memory [60–63]. Treatment of adult Tsc2+/− mice with rapamycin reversed not only the synaptic plasticity of the mice but also the behavioral deficits associated with TSC [60–63]. In other studies in these and other similarly genetically engineered mice, various rapalogs have reversed impaired social interaction and cognition [64]. These results have provided a biological basis for some of the cognitive deficits associated with TSC and a foundation for clinical evaluations of various rapalogs in human TSC [64].
1.11 Other Avenues of Clinical Research
Although this book will principally focus on targeting mTOR/mTORC1 and related signaling elements in malignant diseases, it is clear that the rapalogs have demonstrated the potential to confer major clinical benefit in a wide range of malignant and nonmalignant diseases in just two decades since the discovery of the mechanism of rapamycin. In essence, the identification of rapamycin during the Easter Island expedition in 1964 serendipitously unraveled principal facets about the regulation of cell growth, nutrition, and energy utilization, which may have not been discovered otherwise, at least not for several decades. The serendipitous discovery of rapamycin coupled with highly concerted efforts to identify its target, mTOR, sometimes called the “master regulator,” has resulted in registration of rapamycin and several rapalogs worldwide to treat and prevent refractory cancer, as well as organ rejection following allogeneic transplantation (kidney, liver, heart), autoimmune disorders, and cardiac arterial restenosis, which, in total, affect millions of individuals worldwide each year.
The scope of this chapter is narrow relative to the profound clinical implications, many as of yet unknown, of modulating mTOR/mTORC1. Since mTOR/mTORC1 integrates input from upstream pathways, including insulin, growth factors, and amino acids; senses cellular nutrient, oxygen, and energy levels; and is dysregulated in many important pathological conditions, it is not inconceivable that the rapalogs and novel, versatile small molecule inhibitors of TORC1, TORC2, Akt, PI3K, among other related signaling elements, may be successful at modifying the fundamental pathology of many as of yet untreatable diseases. Further, it is not inconceivable that these agents may be useful for treating several age-associated diseases, including neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, and prevent the effects of premature aging [65–68]. In Alzheimer’s disease, for example, postmortem studies have revealed dysregulation in phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase (PTEN), Akt, ribosomal protein S6 kinase beta-1 (S6K), and mTOR, and aberrant mTOR signaling appears to be closely related to the presence and decreased clearance of soluble amyloid and tau proteins, which aggregate and form two hallmarks of the disease, amyloid plaques and neurofibrillary tangles, respectively [69–73]. Lastly, with regard to aging, decreased TOR activity has been shown to increase lifespan in yeast, and rapamycin has been shown to increase lifespan in mice by several independent groups at the Jackson Laboratory, University of Texas Health Science Center (San Antonio), and the University of Michigan as will be discussed in a later chapter [74–78]. Putative mechanisms involve the role of mTOR in regulating essential nutrients, free radicals, and mitochondrial respiration, and autophagy, among others, but the precise mechanisms that account for these effects are far from clear. Nevertheless, the prospect for developing antiaging therapy that involves targeting mTOR/mTORC1 is not inconceivable [79].
The mTOR signaling pathway has been studied intensively for about 25 years. These research efforts have been facilitated greatly by the serendipitous identification and recent availability of the highly potent and selective mTOR inhibitor rapamycin. Although some important conceptual gaps remain to be filled, the mTOR pathway is now understood at a level of molecular detail that rivals that of any other signaling cascade in mammalian cells. The exceedingly rapid rate of knowledge accumulation in this area stands as a tribute to the combined powers of chemical biology, yeast and Drosophila genetics, and biochemical and genetic studies in mammalian cells. The implications of targeting mTOR and related signaling elements to prevent and treat malignant and nonmalignant disorders with either rapalogs or more versatile small molecule inhibitors are astounding. Nonetheless, the challenges associated with the transition of the rapalogs from the laboratory bench to the oncology clinics have underscored the fact that we still have much to learn about the intricacies of the mTOR pathway itself, as well as the integration of this pathway into the network of signaling cascades that underpins the multitude of genetic subtypes that constitute cancer and other proliferative disorders. However, there is much optimism about making progress in this regard, given the immense headway made to date as discussed in later chapters of this book.
References
1.
Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975;28:727–32.CrossRef
Related posts:
 The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
 Predictive Biomarkers of Response to mTOR Inhibitors
Predictive Biomarkers of Response to mTOR Inhibitors
 mTOR Inhibition Beyond Rapalogs
mTOR Inhibition Beyond Rapalogs
 Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


