Fig. 16.1
Schematic representation of the mitochondrial respiratory chain
The concept of ‘aerobic glycolysis’ suggested by Warburg implies that cancer cells predominantly depend on glycolysis for energy production and to fuel their growing requirement for uncontrolled proliferation and survival in the tumour microenvironment. Even though recent observations suggest that some tumour cells are capable of oxidative phosphorylation, the fact remains that increased dependence of cancer cell on glucose does exist. Various studies have suggested that switching to glycolysis is advantageous to tumour cells for several reasons:
Glycolysis alone generates ATP more rapidly than glycolysis coupled to oxidative phosphorylation, although with a low efficiency (Jezek et al. 2010; Smolkova et al. 2010).
Glycolysis triggers the accumulation of pyruvate and formation of lactate in cancer cells that creates an acidic environment which assist in tumour invasion (Swietach et al. 2007; Wu et al. 2007).
Cancer cells use carbon intermediates of glycolytic pathway for anaplerotic pathways required for synthesis of fatty acids and non-essential amino acids. Moreover, tumours can also metabolise glucose through the pentose phosphate pathway (PPP) to produce ribose 5-phosphate for de novo synthesis of nucleotides and nicotinamide adenine dinucleotide phosphate (NADPH) for all the anabolic process (Marin-Hernandez et al. 2011).
A diminished oxidative phosphorylation also leads to citrate accumulation which further feeds fatty acid and cholesterol synthesis.
The switch from oxidative phosphorylation to glycolysis decreases ROS generation in tumour cells. As ROS also lead to efficient execution of programmed cell death therefore, a diminished ROS production increases the threshold of cancer cell for programmed cell death.
Mitochondrial biogenesis and ETC component synthesis is also impaired in tumour cell and saves energy for other essential metabolic pathways.
16.3 Mechanism of Altered Bioenergetics
The altered metabolism towards aerobic glycolysis in a cancer cell is driven by an increased expression of glucose transporters, glycolytic enzymes and inhibitors of mitochondrial metabolism.
Indeed, cancer cells compensate for lower efficiency of ATP production by glycolysis through the upregulation of glucose import into the cytoplasm by glucose transporters (GLUTs) (DeBerardinis et al. 2008; Hsu and Sabatini 2008). Indeed, this property of increased glucose uptake by malignant cells has been exploited for the visualisation of human tumours using positron emission tomography (PET) with a radiolabelled analogue of glucose (18F-fluorodeoxyglucose, FDG) as a reporter.
Furthermore, increased expression as well as activity of glycolytic enzymes such as hexokinase II, glyceraldehyde 3-phosphate dehydrogenase, lactate dehydrogenase A and glucose-6-phosphate isomerase may determine the outcome of transformation of a normal cell to a malignant cell. Several reports have shown that hexokinase is important to maintain high glucose uptake and catabolic rates in rapidly growing tumours. Hexokinase II (HK II) can associate physically to the outer mitochondrial membrane (OMM) through specific binding to a porin or voltage-dependent anion channel (VDAC) (Mathupala et al. 2009). The VDAC and adenine nucleotide transporter (ANT) provide an advantage to HK II for glucose phosphorylation and allowing it to directly use ATP produced through oxidative phosphorylation. This also allows the cells to maintain a high glycolytic influx by immediately phosphorylating glucose and thereby sustaining the concentration gradient (Mathupala et al. 2006). Interestingly, HK II has also been shown to regulate apoptosis by physically associating with apoptosis-inducing factor (AIF).
Multiple studies have implicated hypoxia-inducible factor 1α (HIF-1α) as a molecular switch between glycolysis and oxidative phosphorylation (Fig. 16.2). As neoplastic cell rapidly proliferates, it outgrows the diffusion of local blood supply that triggers hypoxia and stabilisation of the hypoxia-inducible transcription factor HIF-1α. HIF-1α increases the expression of pyruvate dehydrogenase kinase (PDK) that further inactivates pyruvate dehydrogenase (PDH) enzyme responsible for the conversion of pyruvate into acetyl-CoA. The resultant decrease in acetyl-CoA leads to suppression of Krebs cycle and mitochondrial respiration. The suppression of pyruvate oxidation by HIF-1α also protects cells from production of cytotoxic amounts of ROS. HIF-1α was also shown to stimulate expression of the gene encoding lactate dehydrogenase A, which facilitates conversion of pyruvate into lactate. This effect would further diminish the utilisation of pyruvate by mitochondria and hence suppress mitochondrial respiration (Kim et al. 2006; Papandreou et al. 2006). Additionally, HIF-1α regulates many different genes such as vascular endothelial growth factor, hepatocyte growth factor receptor (c-Met), erythropoietin, transforming growth factor-α, platelet-derived growth factor and glucose transporter GLUT1 and therefore influences cellular activities such as angiogenesis, glycolysis and cell survival (Semenza 2003).
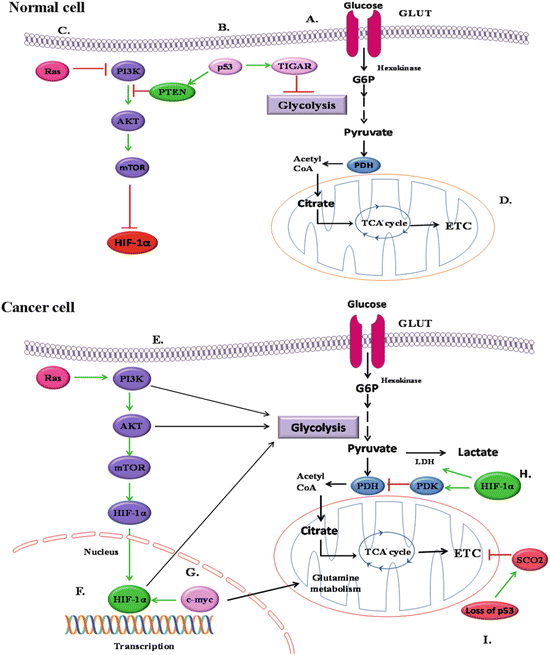
Fig. 16.2
Alterations in mitochondrial bioenergetics are regulated by different signalling events: (a) uptake of glucose through glucose transporter GLUT. (b) In normal cells, tumour suppressor gene p53 activates PTEN which further inhibits the PI3/Akt/TOR pathway, thereby leading to HIF-1α inactivation. p53 also activates TIGAR (TP53-induced glycolysis and apoptosis regulator) which inhibits the process of glycolysis. (c) Wild-type Ras also inactivates HIF-1α by inhibiting PI3/Akt/TOR signalling pathway; (d) glycolysis leads to oxidative phosphorylation in normal cells. (e) In cancer cells, oncogenic activation of Ras promotes the stabilisation of HIF-1α by activating PI3/Akt/TOR signalling events; (f) stabilisation and activation of HIF-1α leads to enhanced glycolysis. (g) Oncogenic transcription factor, Myc, activates HIF-1α and also glutamine metabolism. (h) Stabilised and activated HIF-1α leads to the activation of lactate dehydrogenase (LDH) which converts pyruvate into lactate. HIF-1α also activates enzyme pyruvate dehydrogenase kinase (PDK) which inhibits the activity of enzyme pyruvate dehydrogenase (PDH) and therefore suppresses TCA cycle and oxidative phosphorylation. (i) Mutation in p53 causes downregulation in the expression of cytochrome c oxidase 2 (SCO2) gene and inhibits the process of oxidative phosphorylation
Moreover, a number of well-established mutated tumour suppressors and activated oncogenes play critical roles in the regulation of glycolytic fuelling and thereby may suppress or promote uncontrolled proliferation (Fig. 16.2) (DeBerardinis et al. 2008; Jones and Thompson 2009). The oncogenic mutation of Ras activates PI3K/Akt/mTOR pathway which promotes glycolysis through the induction of HIFs. The oncogenic transcription factor, Myc, also cooperates with HIF to activate the transcription of glycolytic enzymes such as hexokinase or phosphofructokinase as well as lactate dehydrogenase A and PDK1. Myc also regulates the genes associated with glutamine metabolism. The high rate of glycolysis and glutaminolysis has been observed in Myc-induced hepatic cancer (Hu et al. 2011). Similarly, tissue-type transglutaminase (TG2), a proinflammatory protein whose expression is frequently upregulated in multiple drug-resistant and metastatic tumour types, was recently shown to reprogramme glucose metabolism in epithelial mammary cells due to increased expression of HIF-1 even under normoxic conditions (Kumar et al. 2014). Another important signalling event is PI3K/Akt/mTOR pathway which increases the uptake of glucose and stimulates anaerobic glycolysis in tumour cells (Jones and Thompson 2009). Akt regulates not only the localisation of the glucose transporter to the cell surface but also phosphorylation and activity of glycolytic enzymes such as hexokinase and phosphofructokinase. Binding of HK II to VDAC in the outer mitochondrial membrane is enhanced by overexpression of Akt due to negative regulation of the activity of glycogen synthase kinase-3-β (GSK-3-β) (Robey and Hay 2009). The two important events regulated by PI3K are its antagonist effect on PTEN and upregulation of mTOR pathway. Both of these events further lead to activation of glycolysis and induction of HIF-1α even under normoxic conditions.
In contrast to oncogenes, the activation of p53 tumour suppressor gene leads to an increase in mitochondrial respiratory activity and decrease in glycolysis. Loss of p53 function actually downregulates the expression of gene encoding the synthesis of cytochrome c oxidase 2, the product of which is required for the assembly of cytochrome c oxidase of the mitochondrial respiratory chain (Buchwald et al. 1991). Therefore, mutations in p53 can suppress mitochondrial respiration as a result of cytochrome c oxidase deficiency and facilitate the shift of cellular energetic metabolism towards glycolysis. A second target of p53 is TIGAR (TP53-induced glycolysis and apoptosis regulator) which inhibits the glycolysis by decreasing levels of fructose-2,6-bisphosphate, a potent stimulator of glycolysis and inhibitor of gluconeogenesis (Bensaad et al. 2006) (Fig. 16.2).
16.4 Mitochondrial Mutations and Cancer
Additional support to mitochondrial involvement in cancer pathogenesis was provided by the complete sequencing of mitochondrial genome and discovery of oncogenic mutations in mitochondrial metabolic enzymes such as fumarate hydratase (FH), succinate dehydrogenase (SDH) and isocitrate dehydrogenase (IDH). Mutations in both the tricarboxylic acid cycle (TCA) enzymes such as succinate dehydrogenase (SDH) and fumarate hydratase (FH) lead to the accumulation of their substrates succinate and fumarate respectively. The increased succinate and fumarate levels result in stabilisation and activation of HIF-1α (Kurelac et al. 2011; Wallace 2012). The stabilised HIF-1α translocates into the nucleus and causes a shift in energy metabolism towards glycolysis in cancer cells by multiple pathways as mentioned earlier (Frezza et al. 2011). SDH complex II of ETC is also considered as a classical tumour suppressor, and mutations in this enzyme may affect the mitochondrial respiration (Fogg et al. 2011). A defect or mutation(s) in this specific metabolic enzyme has been also reported in familial predisposition to benign tumours. Similarly, various studies have reported that germ line mutations in tumour suppressor gene FH are associated with leiomyoma and renal cancer (Martinez-Mir et al. 2003). NADP (nicotinamide adenine dinucleotide phosphate)-dependent isocitrate dehydrogenase 2, (IDH2), is another enzyme of TCA cycle which catalyses the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (α-KG) and generate NADPH. Somatic mutations of IDH1 and IDH2 have been found in gliomas, astrocytomas, chondromas and acute myeloid leukaemia (AML) (Green and Philip 2010; Yan et al. 2009). These mutations have been associated with a decrease in the levels of cytoplasmic α-KG and inhibition of prolyl hydroxylase activity which in turn lead to stabilisation of HIF-1α. Interestingly, mutant IDH1 and IDH2 also convert α-KG to 2-hydroxyglutarate (2-HG), which is hypothesised as an ‘oncometabolite’. Mutations in IDH1 and IDH2 may also alter the cellular redox status by utilising NADPH for its activity. The diminished NADPH further inhibits the activity of glutathione peroxidase and increases H2O2 levels. Hence, the decreased NADPH affects redox status and thereby favours the proliferation and tumorigenesis (Wallace 2012).
16.5 Targeting Metabolic Reprogramming in Cancer Cell
The inherent differences in glucose metabolism between a normal and a cancer cell have been extensively utilised for designing therapeutic strategies against cancer. The concept of development of antineoplastic regimens is based on killing the tumour cell selectively by apoptosis without affecting the normal cell. Multiple studies have suggested that hyperglycolytic state of a tumour cell makes mitochondria less susceptible to apoptosis by interfering with permeabilisation of outer mitochondrial membrane, a regulatory step in mitochondria-mediated apoptotic pathway (Weinberg and Chandel 2009; Jourdain and Martinou 2009).
The inhibitory effect of glucose deprivation in carcinogenesis is attributed to either death receptor-mediated apoptosis (Muñoz-Pinedo et al. 2003) or induction of ROS selectively in cancer cells (Spitz et al. 2000; Simons et al. 2009).
Administration of glucose analogues has also been investigated for their ability to abolish carcinogenesis. Although it is not possible to establish a complete glucose-deprived environment in vivo, it is promising to treat animals or patients with glucose analogues. 2-Deoxy-D-glucose (2DG) is a non-metabolisable glucose analogue that competes with glucose for transport and further phosphorylation by hexokinase II to enter into glycolysis, thus creating a state of glucose deprivation. The administration of 2DG in animal model showed minimal toxicity except at a very high dose. However, later studies have shown that 2DG activates prosurvival mechanism in cancer cells (Zhong et al. 2009). Accordingly, the success of 2DG alone in treatment of cancer has been challenged but has provided promising outcomes in combination therapies with cisplatin, L-buthionine-[S,R]-sulfoximine (BSO) and ionising radiation (Simons et al. 2010). In addition to promoting apoptosis, 2DG has been shown to have antiangiogenic effect both in vitro and in vitro (Merchan et al. 2010).
Another beneficial strategy to combat carcinogenesis is by inhibition of glucose transport. Phloretin, an apple flavonoid, interferes with GLUT2 transport mechanism and inhibits cancer growth by inducing apoptosis in HT29 cells (Park et al. 2007) and HepG2 liver cancer cells (Wu et al. 2009).
Similarly the anticarcinogenic potential of resveratrol, a phytoalexin found in grapes, has been linked to reduced glucose uptake and lactate production in ovarian cancer cells (Kueck et al. 2007). These findings suggest that glycolysis or glucose transport inhibitors mimic glucose deprivation and could be potential complement to the existing cancer therapies.
3-Bromopyruvate (3-BrPA) is a derivative of pyruvate that has been shown to abolish advanced stage hepatic cancer growth without harming normal cells both in vitro and in vitro (Geschwind et al. 2002; Ko et al. 2004). 3-BrPA inhibits hexokinase II activity by disrupting its association with AIF eventually leading to cell death (Chen et al. 2009). Recently, the FDA has accepted an investigational new drug (IND) application for 3-BrPA for Phase I clinical trial in liver cancer.
Annonaceous acetogenins, long chain fatty acid derivatives extracted from Annona muricata, exerts multiple anticancer activities in pancreatic cancer. This compound has been reported to inhibit glucose uptake, HK II and LDH-A mediated by NF-κB-dependent regulation of HIF-1 (Torres et al. 2012). The plant phenols fisetin, myricetin, quercetin, apigenin, genistein, cyanide, daidzein, hesperetin, naringenin and catechin also inhibit glucose uptake by tumour cells and thereby exert cancer chemotherapeutic effect (Park 1999). Genistein binds the glucose transporter on the external face whereas quercetin interacts with the internal face (Perez et al. 2011).
The inhibition of HK II activity and destabilisation of its binding to VDAC not only affects the cancer cell energetics but also induces apoptosis by disrupting mitochondria and releasing cytochrome c. Methyl jasmonate, produced by rosemary, olive or ginger, binds to HK II and thereby interferes its binding with VDAC in cancer cell (Cohen and Flescher 2009).
Fatty acid synthetase is also considered as an attractive target for cancer therapy. In this context cerulenin [(2S,3R)]-2-3-epoxy-4-oxo-7,10-dodecadienoylamide, which is a fungal metabolite and an inhibitor of FASN, has been observed to dissociate HK II from AIF in ZR-75-1 human breast cancer cells (Jeong and Yoo 2012). Another natural occurring flavonoid, Oroxylin A isolated from Chinese traditional medicinal plant Scutellaria baicalensis Georgi, has been shown to cause detachment of HK II from mitochondrial membrane in human breast cancer cell lines (Wei et al. 2013).
As reported, the preferential conversion of glucose to lactate rather than pyruvate in tumour cell is governed by the enhanced activity of LDH-A and downregulation of PDH. The phosphorylation status of PDH is further regulated by PDK I which has been found to be overexpressed in cancer (Raimondi and Falasca 2011). Redirecting pyruvate towards mitochondria by inhibiting either LDH-A and PDK I alone or both has been explored as an attractive therapeutic strategy. Oxamate, an inhibitor of LDH-A, has been reported to suppress the cell proliferation in a dose- and time-dependent manner in nasopharyngeal cancer cell lines. The induction of apoptosis by oxamate has also been attributed to its ability to enhance mitochondrial ROS generation. In addition, oxamate also increased the sensitivity of nasopharyngeal cancer cells to ionising radiations both in vivo and in vitro (Zhai et al. 2013). The use of shRNAs to silence LDH-A gene resulted in suppression of growth in breast cancer cell lines as well as xenograft mouse model (Wang et al. 2012). The orphan drug dichloroacetate (DCA) which had the ability to inhibit cancer growth selectively was reported to be a PDK I inhibitor and has led to the reduction in tumour growth by inducing ROS, activating caspase-3 and apoptosis in multiple myeloma cells (Fujiwara et al. 2013). DCA has crossed Phase II clinical trials for treating glioblastoma (Michelakis et al. 2010). Due to the bioavailability concern of orally administered DCA, Mito-DCA were designed to selectively target this drug to mitochondria. The observations with Mito-DCA suggested increased efficiency and selectivity of this drug which alter the glucose metabolism only in the cancer cell while sparing the normal cells. Further experiments revealed that Mito-DCA causes a reduction in mitochondrial membrane potential (Δψm) that might ease the release of cytochrome c and subsequent cell death (Pathak et al. 2014).
Another well-known chemopreventive agent, curcumin, has also been reported to target mitochondrial metabolism and function in a variety of cancer model (Jurenka 2009; Teiten et al. 2010). Similar to Mito-DCA, mitochondrial-targeted curcuminoids (mitocurcuminoids) have been synthesised to circumvent the low bioavailability problem of curcumin both at tissue and plasma levels. ESI-MS analysis showed significantly higher uptake of the mitocurcuminoids in mitochondria as compared to curcumin in breast cancer cells. These mitocurcuminoids also have been shown to exhibit significant cytotoxicity to different cancer cell lines with minimal effect on normal mammary epithelial cells (Reddy et al. 2014).
Aberrant expression of mitochondrial protein frataxin leads to development of Friedreich ataxia. Overexpression of frataxin has been shown to induce oxidative metabolism, increased aconitase activity, mitochondrial membrane potential, cellular respiration and ATP content in several colon cancer cell lines. The induction of oxidative metabolism by frataxin was attributed to its ability to stimulate iron/sulphur clusters synthesis in mitochondria. Furthermore, frataxin also suppressed the tumour formation in mouse xenograft model, thus acting as a tumour suppressor (Schulz et al. 2006). Frataxin has also been reported to be overexpressed in response to hypoxic condition in tumour cells mediated by HIF (Guccini et al. 2011).
The mitochondrial ETC itself is instrumental in apoptosis induction, and deficiencies in ETC produced by mutations in mitochondrial genome (mtDNA) or in some nuclear-coded components predispose to carcinogenesis (Kwong et al. 2007). In this regard, dietary constituents have been reported to inhibit the process of carcinogenesis by altering mitochondrial metabolic pathway and stimulating ETC. n-3 Polyunsaturated fatty acids (n-3PUFAs), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been shown to alter the mitochondrial membrane lipid profile to make it more susceptible to apoptosis (Sharma et al. 2013). These have been reported to inhibit glucose oxidation by glycolysis by stimulating ETC (Barrera 2012; Merendino et al. 2013).
Related posts:
 Prevalence of Gastrointestinal Cancers in India
Prevalence of Gastrointestinal Cancers in India
 Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
 Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
 Apoptosis Pathways in Chronic Lymphocytic Leukemia: Role of the Microenvironment and Therapeutic Strategies
Apoptosis Pathways in Chronic Lymphocytic Leukemia: Role of the Microenvironment and Therapeutic Strategies
 Role of Surfactants in Regulation of Cancer Growth
Role of Surfactants in Regulation of Cancer Growth
 Personalized Therapeutic Strategies for Epithelial Ovarian Cancer
Personalized Therapeutic Strategies for Epithelial Ovarian Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


