Fig. 14.1
Mechanisms of hypoxia-inducible factor (HIF)-1α regulation under aerobic and hypoxic conditions. VHL von Hippel-Lindau
14.3 The Role of HIF-1 in Tumorigenesis
Data from genome-wide chromatin immunoprecipitation coupled to next-generation high-throughput screening (ChIP-seq) studies suggest that HIF directly regulates over 800 genes involved in many critical aspects of cancer biology (Schödel et al. 2011). In response to hypoxia, HIF-1 induces expression of growth factors such as insulin-like growth factor-2 (IGF-2) and transforming growth factor-α (TGF-α). These growth factors bind to their cognate receptors to promote cell proliferation/survival, as well as the expression of HIF-1α itself through autocrine-signalling pathways.
This growth is supported by HIF-1 dependent expression of erythropoietin (EPO) and vascular endothelial growth factor (VEGF), which facilitates erythropoiesis and angiogenesis to increase the oxygen carrying capacity and vascular density respectively. Numerous HIF-1 dependent metabolic reprogramming mechanisms also occur. Experimental evidence from HIF-1α knockout mice unequivocally demonstrated that HIF-1 up-regulates expression of all glucose transporters (GLUT1, GLUT2), glycolytic enzymes (GAPDH, LDHA), and glycogen storage enzymes (GYS1, GBE1). It also orchestrates a subunit switch in cytochrome c oxidase to increase the efficiency of the electron transport chain, enabling continued respiration in conditions of moderate hypoxia without increased levels of reactive oxygen species (ROS). HIF-1 further up-regulates the expression of pyruvate dehydrogenase kinase 1 (PDK1), which phosphorylates and inactivates pyruvate dehydrogenase (PDH) under severe hypoxia to inhibit oxidation of pyruvate into acetyl-CoA for entry into the TCA cycle. To compensate for the reduced flux of glucose to citrate, reductive carboxylation of glutamine is used to generate cytosolic citrate for fatty acid synthesis. Collectively, these changes facilitate the switch from mitochondrial oxidative metabolism to anerobic glycolysis to maintain an undifferentiated state under hypoxic conditions (Semenza 2013).
HIF-1 also promotes epithelial-mesenchymal transition by inducing transcription of gene coding repressors (ID2, ZEB2) which mediates the loss of E-cadherin and other proteins that contribute to maintaining the cellular cytoskeleton and cell-cell adhesion. It also induces transcription of LOX and matrix metalloproteinases (MMPs), which augments hypoxia-induced invasion of tumour cells and formation of metastatic lesions in animal models.
14.4 HIF-1 as a Therapeutic Target in Cancer
HIF-1 over-expression in human cancer is associated with poor survival. Immunohistochemical analyses of oropharyngeal (Aebersold et al. 2001) and esophageal squamous-cell carcinoma (Tzao et al. 2008), as well as laryngeal (Schrijvers et al. 2008), gastric (Griffiths et al. 2007; Takahashi et al. 2003), pancreatic (Sun et al. 2007), colorectal (Rajaganeshan et al. 2008), and rectal carcinoma (Rasheed et al. 2009) biopsies also revealed a positive correlation between HIF-1 overexpression, radiotherapy resistance, and increased patient mortality.
The importance of HIF-1 in tumour growth is emphasized by experimental data in which overexpression of HIF-1α in HCT116 colorectal cancer (Ravi et al. 2000) and PCI-10 pancreatic cancer cells (Akakura et al. 2001) increased vessel density and tumour growth respectively. Critically, previous studies have demonstrated that HIF-1α null mutations severely impede tumour growth by reducing expression of glycolytic enzymes, with tumours unable to grow beyond 2 mm3 unless supported by neovascularization mediated by hypoxic induction of HIF-1 (Ma and Adjei 2009; Ryan et al. 1998). In contrast to VEGF inhibitors, HIF-1 inhibitors were also shown to decrease breast cancer cell metastasis in mouse orthotopic transplantation models (Zhang et al. 2012), and sensitize tumours to radiotherapy (Moeller et al. 2004). Additionally, anti-sense blockade of HIF-1α expression in gastric carcinoma cells reduced VEGF production in response to COX-2 overexpression (Huang et al. 2005), with the introduction of HIF-1α siRNA into a glioma cell line down-regulating MMP-2/MMP-9 to suppress cell migration and invasion into adjacent normal tissue (Fujiwara et al. 2007). The siRNA also selectively prevented hypoxia-induced treatment resistance (Sullivan et al. 2008).
Since HIF-1 mediates multiple tumour survival mechanisms, and its overexpression arises not only due to the actions of bioactive lipid mediators, viral infections, and intratumoral hypoxia, but also through oncogene gain-of-function (Giatromanolaki et al. 2004) and tumour suppressor gene (Maxwell et al. 1999) loss-of-function mutations, HIF-1 may represent a final common pathway in cancer pathogenesis. Therefore, HIF-1 is an attractive target for cancer therapy.
14.4.1 Pharmacological Targets in the HIF-1 Pathway
Small-molecule inhibitors of HIF-1 are highly desirable due to its central role in tumorigenesis. Broadly, small-molecules may inhibit HIF-1 by decreasing its protein levels, or by preventing its dimerization, DNA-binding, or transactivation (Fig. 14.2). Sorafenib, and P3155 have been found to decrease HIF-1 synthesis (Liu et al. 2012; Manohar et al. 2011), with systemic digoxin treatment in tumour-bearing mice blocking lymphatic metastasis to axillary lymph nodes (Schito et al. 2012). Other examples include KC7F2 and PX-478, which inhibits translation via a VHL- and p53-independent mechanism (Koh et al. 2008; Narita et al. 2009), and the RNA antagonist EZN-2968, which inhibits expression of HIF-1α mRNA (Greenberger et al. 2008). Notably, PX-478 and EZN-2968 cause dose-dependent reductions in levels of HIF-1α, VEGF secretion, and tumour size in xenograft models, and were both well tolerated in Phase I clinical trials (Greenberger et al. 2008; Jeong et al. 2014; Koh et al. 2008). Further modulators identified to date include geldanamycin, which reduces HSP90 binding to HIF-1α to destabilize folding and increase proteasomal degradation (Isaacs et al. 2002), and the mTOR inhibitor rapamycin. Indirect actions through upstream or downstream signaling pathways however cause many side effects. The development of novel specific HIF-1 inhibitors is thus needed.
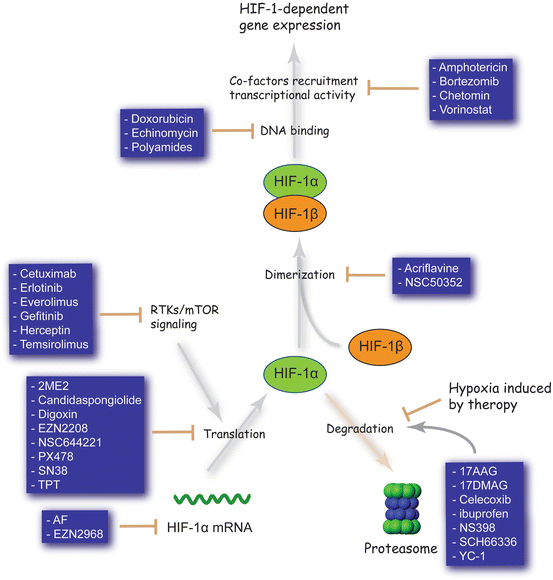
Fig. 14.2
HIF-1 inhibitors. These are meant to be illustrative, rather than comprehensive, examples
To identify HIF-1 inhibitors with greater selectivity, there are on-going efforts to target the protein-protein interactions. Traditionally considered to be an undruggable target due to its weak and transient nature, preventing HIF-1 heterodimerization is a challenging but potentially rewarding strategy. The small-molecule acriflavine, which inhibits HIF-1α and HIF-1β dimerization, acts as a radiosensitizer and potently inhibited prostate cancer xenograft growth and breast cancer metastasis to the lungs in mouse models (Lee et al. 2009b; Lim et al. 2012). The new peptide cyclo-CLLFVY may hold more promise in this regard. Using a high-throughput genetically encoded screening platform, cyclo-CLLFVY was found to bind to the PAS-B domain of HIF-1α. It mediated selective inhibition of the HIF-1 mediated cellular response by disrupting dimerization in MCF-7 and U2OS cell lines (Miranda et al. 2013). However, cyclic peptides have difficult pharmacokinetics, making the transition to clinical use challenging.
In an alternate approach, echinomycin and anthracycline antibiotics may be used to inhibit HIF-1: DNA-binding via DNA intercalation at the hypoxia response element (HRE) (Kong et al. 2005; Lee et al. 2009a). DNA intercalators however show limited sequence specificity, thus causing off-target effects. Indeed, echinomycin cannot be used in cancer chemotherapy due to its secondary action on Sp1 (5′-CCGCCC-3′), which ultimately increases HIF-1α expression under normoxic conditions. A more promising strategy may be to target the DNA-binding domain of HIF-1α (unpublished work).
14.4.2 Limitations of Previous Approaches
Numerous cancer chemotherapeutic agents target angiogenesis in tumours to reduce primary tumour growth. Response to anti-VEGF therapy has however been poor, as intratumoral hypoxia arising from impaired angiogenesis causes HIF-1 dependent metastasis and expansion of cancer stem cell pools (Conley et al. 2012). Hence targeting HIF-1 activity may abrogate the compensatory pathways required for cancer cell survival.
Indeed, it has been shown that bevacizumab increases intratumoral hypoxia and expression of HIF-dependent genes in glioma xenografts, and that inhibition of HIF-1α by topotecan significantly reduces tumour cell proliferation (Rapisarda et al. 2009). Critics, however, argue that topotecan has failed to show any anti-cancer effects in previous clinical trials. The key distinction is that HIF inhibitors were previously used as cytotoxic agents and administered episodically at the maximum tolerated dose, whereas continuous inhibition of HIF activity requires frequent administration at lower doses. This is supported by evidence from a recently completed pilot study involving 16 patients with advanced cancer and biopsy-proven HIF-1α overexpression, where topotecan resulted in decreased tumour blood flow in seven out of ten patients as measured by dynamic contrast enhanced magnetic resonance imaging (DCE-MRI), and a loss of HIF-1α expression in four out of seven patients (Kummar et al. 2011). Further research is needed to determine the optimal dosing schedule for chronic use of HIF inhibitors as anti-cancer agents.
14.4.3 Single Agent Versus Combination Therapy
There is overwhelming evidence for the use of HIF-1 inhibitors in combination therapy. Hypoxia promotes selection of a treatment resistant phenotype by promoting transcription of genes involved in multidrug resistance (MDR-1, ABC transporters), and down-regulating genes involved in DNA mismatch repair. However, tumour cells have heterogeneous expression of HIF-1, thus conceptually, HIF-1 inhibitors used as a single agent may be ineffective. In support of this hypothesis, experimental evidence from combination treatment with rapamycin and LBH589, a mTOR and HDAC inhibitor respectively, had significantly greater anti-angiogenic and anti-tumour activity in PC3 and C2 cell lines in vivo compared with single agents (Verheul et al. 2008). Similarly, addition of low dose daily topotecan – a HIF inhibitor – to bevacizumab significantly inhibited tumour growth and reduced microvessel-density relative to mice treated with topotecan or bevacizumab alone in U251-HRE xenografts (p < 0.01) (Rapisarda et al. 2009). It is plausible that the increase in cytotoxic activity of combination therapy is due to inhibition at different levels of HIF-1α regulation, or by inhibition of other oncogenic pathways.
The role of HIF inhibitors on the effects of radiotherapy for hypoxic tumours is less clear. HIF inhibitors given prior to treatment may suppress the effects of radiotherapy because its anti-angiogenic effects increases the radio-resistant hypoxic fraction. However, pre-clinical studies have also demonstrated an inverse correlation between hypoxia and local tumour control after irradiation. Examples include the use of HIF inhibitors BAY-84-7296 and YC-1 in combination with radiotherapy, both of which significantly enhanced the effects of radiotherapy against tumour xenografts if given after the radiation had been delivered. This may be due to hypoxia-induced expression of pro-angiogenic factors, which promote survival of endothelial cells following radiation, or through protection of cancer stem cells. Hence the timing of the treatment regime regarding the use of HIF inhibitors in combination with radiotherapy is critical (Harada et al. 2009).
14.4.4 Contraindications to Treatment with HIF-1 Inhibitors
One potential caveat is that patients with anemia or severe ischemic cardiovascular disease may experience exacerbation of their condition during treatment. In support of this hypothesis, post-hoc analysis from the PROactive randomized controlled trial showed the pioglitazone – an inhibitor of autocrine and paracrine angiogenesis through mitochondrial stabilization of HIF-1 – was associated with critical limb ischemia and an increased hazard for surgical or percutaneous lower extremity revascularization in diabetic patients (Dormandy et al. 2009). However, this is only a relative contraindication for its use.
14.5 Exploring HIF-Independent Strategies
Although the most studied cellular response to hypoxia is mediated by the transcription factor HIF-1, several HIF-independent mechanisms have also been implicated in hypoxic adaptation. These include the unfolded protein response (UPR) and the mTOR signalling pathway, which curtail oxygen consumption by energy expensive processes such as protein synthesis. Given their pivotal role in cellular adaptation to hypoxic stress, they are of considerable interest as therapeutic targets.
14.5.1 The Unfolded Protein Response (UPR)
Hypoxia, through the lack of oxygen to act as the terminal electron acceptor in the redox relay for disulphide bond formation, impairs protein folding in the ER. Accumulation of unfolded proteins within the ER activates the UPR, which mediates a global reduction in transcription and translation through the PERK-eIF2A-ATF4, IRE1-XBP1, and ATF6 signalling pathways. Clinical data suggests that all three branches of the UPR are up-regulated in human cancers. ATF4 over-expression has been found in brain, breast, cervical, and skin cancers as compared to adjacent normal controls (Bi et al. 2005). Levels of spliced XBP1 and ATF6 mRNA were also elevated in hepatocellular carcinoma samples (Shuda et al. 2003). Critically, transformed mouse embryo fibroblasts (MEFs) derived from PERK- and XBP1- knockout mice exhibit reduced clonogenic survival after hypoxic stress, as well as impaired tumour growth (Bi et al. 2005; Romero-Ramirez et al. 2004). Similar results were observed following transfection of short hairpin RNA (shRNA) against ATF4 into HT1080 human fibrosarcoma cells (Ye et al. 2010). These studies support the concept of targeting the UPR to inhibit tumour growth.
Two therapeutic strategies are currently being pursued. One approach is to inhibit the UPR by targeting its components PERK, ATF4, and IRE1. Salicylaldehyde analogues and the small-molecule GSK2606414A have recently been identified as potent and selective inhibitors of IRE1 and PERK respectively (Axten et al. 2012; Volkmann et al. 2011). The antidiabetic biguanides, such as metformin, also inhibit production of UPR transcription activators XBP1 and ATF4 to induce cell death under conditions of glucose deprivation (Saito et al. 2009). Significantly, metformin re-profiled to treat cancer would have an immediate clinical impact. A second approach seeks to exacerbate ER stress in order to overwhelm the UPR, shifting the balance in favour of pro-apoptotic functions. In support of this therapeutic strategy, ER stressors such as thapsigargin or the clinically approved proteasome inhibitor bortezomib exhibit hypoxia-selective cytotoxicity (Fels et al. 2008). Chloroquine, which inhibits autophagy to aggravate ER stress, has also been shown to reduce the fraction of viable hypoxic tumour cells and increase tumour response to radiation (Rouschop et al. 2010). In summary, the UPR is an important mediator of the hypoxic microenvironment, and therapeutic strategies to inhibit the UPR shows promising anti-tumour effects.
14.5.2 The mTOR Signalling Pathway
mTOR senses cellular energy, nutrient, and oxygen levels to regulate cell growth and survival. Under hypoxic conditions, mTOR complex 1 (mTORC1) kinase activity is inhibited by activation of the tuberous sclerosis protein I (TSC1)-TSC2 complex through activation of AMP-activated protein kinase (AMPK) or transcriptional up-regulation of regulated in development and DNA damage responses 1 (REDD1). Similarly, the hypoxia-inducible pro-apoptotic protein BNIP3 is reported to inhibit mTORC1 by direct binding to RAS homologue enriched in brain (RHEB). The resulting suppression of mTORC1 causes hypophosphorylation of 4E-BP1, which leads to its increased association with the cap-binding protein eIF-4E and inhibition of cap-dependent translation. The extent of this inhibition depends on the duration and severity of hypoxia.
It is hypothesized that hypoxia has a dual-role in tumorigenesis. In small, early stage tumours, moderate hypoxia (~1 % O2) should suppress tumour growth through negative regulation of mTORC1. Accumulating evidence, however, suggests that the mTOR signalling network is frequently dysregulated in human cancers. This is corroborated by experiments in two independent p53 null fibroblast lines, whereby the resulting decrease in REDD1 expression reduced sensitivity to oxidative stress (Ellisen et al. 2002). Thus, it is plausible that hypoxia-driven inhibitory mechanisms for down-regulating the mTOR pathway are impaired in tumour cells (Schneider et al. 2008).
Several studies have investigated the activity of pharmacological mTOR inhibitors in hypoxic cells. Rapamycin and its derivatives (rapalogues) crosslink with the immunophilin FK506-binding protein (FKBP12) to form a complex which binds the FKBP-rapamycin binding domain of mTOR to inhibit mTORC1 allosterically. It further inhibits HIF-1α accumulation to mediate hypoxia-selective anti-proliferative effects. The success of rapalogues in clinical trials have however been limited. This may be due to stimulation of the anti-apoptotic PI3K-AKT pathway, as a result of mTORC1 inhibition and up-regulation of IGF-1R signalling. In support of this hypothesis, tissue samples from patients with colon or breast cancer after 4 weeks of treatment with rapalogues had higher levels of activated AKT compared to pre-treatment samples (O’Reilly et al. 2006). Significantly, rapalogue treatment also lead to MAPK activation through a PI3K-dependent feedback loop (Carracedo et al. 2008). Hence dual mTOR/PI3K inhibitors may be a more effective therapeutic approach.
mTORC1 suppression by severe hypoxia (~0.1 % O2) in late stage tumours, however, may be an adaptive response in face of energy limitations which paradoxically favours hypoxic cell survival. The consequences of mTOR inhibition are thus difficult to predict without appropriate patient stratification. More alarmingly, there is significantly interplay between the mTOR, UPR, and HIF-1 signalling pathways. Given its central role in other aspects of cell growth and metabolism, mTOR presents as a less well-defined opportunity to target hypoxic cell survival.
14.6 Perspectives
Therapeutic opportunities in targeting tumour hypoxia have yet to be fully realized. This is despite identification of numerous molecular targets, including HIF-1, the UPR, and the mTOR signalling pathway. From a drug discovery point of view, a better understanding of the interplay between these molecular targets are needed to design drugs with greater selectivity. Improvements in drug delivery systems are also needed to facilitate selective tumour targeting. The high rates of attrition in the pipeline, however, remains the most important issue to be addressed. Thus drug development in the future should consider using re-profiling techniques to reduce failure due to toxicity. Moreover, clinical efficacy may be improved by developing methods of identifying tumour hypoxia, facilitating the selection of patients who will benefit most from hypoxia-modifying treatment.
14.6.1 Teaching an Old Drug New Tricks
Retrospective studies estimate that it takes an average of 15 years and over USD $1 billion to bring a single drug candidate onto the market. Traditional drug discovery using high-throughput screening (HTS) in a chemical space with over 1060 compounds is not only time-consuming, but also expensive. More alarmingly, new drug approvals by the US Food and Drug Administration (FDA) have plunged by 40 % since 2005 despite a concomitant doubling of investment in pharmaceutical research and development. This trend is unsustainable and there is an urgent need to solve the high rates of candidate drug attrition. One solution is to identify new uses for old drugs.
The Nobel laureate James Black famously once stated, “The most fruitful basis for the discovery of a new drug is to start with an old drug.” In drug re-profiling, the molecular mechanisms of existing drugs are re-examined for novel therapeutic indications based on the concept of polypharmacology, where the average drug acts on five different targets (Overington et al. 2006). Screening of the John Hopkins Drug Library, a collection of small-molecules which have either been approved by the FDA or have entered phase II clinical trials, led to the serendipitous discovery of cardiac glycosides (Huafeng Zhang et al. 2008), anthracyclines (Lee et al. 2009a), and acriflavine (Lee et al. 2009) as inhibitors of HIF-1 synthesis, transcription, and dimerization respectively. Mitoxantrone was subsequently found to inhibit HIF-1α mRNA protein translation in a topoisomerase-II independent manner (Toh and Li 2011). In total, it is estimated that nearly 9,000 drugs are off-patent and available for such investigation. Critically, these drugs have known safety and pharmacokinetic profiles. This represents a significant cost advantage over traditional drug development, where more than 90 % of candidate molecules fail clinical testing due to safety concerns. They are also clinically used, enabling immediate commencement of phase II clinical trials for efficacy, and the move to clinic potentially rapid.
14.6.2 Targeted Drug Delivery
Nanopharmaceuticals are the next chapter in the fight against cancer. Early clinical trials with camptothecin – a potent dual inhibitor of topoisomerase I and HIF-1α – demonstrated high anti-tumour activity, but severe toxicity lead to discontinuation of the drug’s development. This failed, but otherwise efficacious drug, may be rescued by shielding it in nanoparticles to improve its pharmacokinetics and safety profile. CRLX101 is a new nanopharmaceutical which contains a cyclodextrin-containing polymer conjugated to camptothecin. It is hypothesized that CRLX101 preferentially targets tumour cells by exploiting the large pores and fenestrations in tumour neovasculature. Sustained release of the drug, as opposed to a burst release followed by a rest period, also reduces the likelihood of developing drug resistance. In a combined phase 1/2 clinical trial, CRLX101 showed encouraging safety, pharmacokinetics, and efficacy – 64 % of patients treated at the maximum tolerated dose of 15 mg/m2 bi-weekly achieved stable disease, with a median progression-free survival time of 3.7 months (Weiss et al. 2013). Phase 2 clinical development across multiple tumour types is ongoing.
Hypoxia-activated prodrugs are also highly desirable. The prodrugs are inactive when administered, and converted in a two-step process in vivo. Firstly, one-electron (1e) reduction of the relatively non-toxic prodrug creates an oxygen-sensitive intermediate. The intermediate undergoes spontaneous conversion into the active drug under hypoxic conditions, but is rapidly re-oxidized into the inert prodrug in the presence of oxygen. This futile redox cycle suppresses formation of the initial prodrug radical in oxic conditions, facilitating hypoxia-selective cytotoxicity. There are currently four classes of hypoxia-activated prodrugs in development, of which the most promising candidate is TH-302 – a 2-nitroimidazole-triggered bromo analogue of the FDA approved DNA-alkylating agent ifosfamide. Initial studies of TH-302 with doxorubicin in advanced soft-tissue sarcoma demonstrated a high objective response rate, and a median overall survival (OS) of 17.5 months (Chawla et al. 2011). More recently, a randomized controlled trial involving 214 patients demonstrated a higher response rate to TH-302 in combination with gemcitabine as opposed to gemcitabine alone in advanced pancreatic cancer, with increases in median progression-free survival (PFS) of 5.5 months and 3.6 months respectively (Borad et al. 2012). Results from ongoing phase III trials in this setting are eagerly anticipated. Nevertheless, significant challenges remain in the development of hypoxia-activated prodrugs. One complicating factor is that many prodrugs may be activated by oxygen-insensitive two-electron (2e) oxidoreductases, resulting in cytotoxicity independent of hypoxia. Another key limitation is that hypoxic regions within tumours are frequently necrotic, and thus lack the enzymes and cofactors needed to reduce the prodrug. The use of hypoxia-regulated gene therapy is an intriguing solution to the problem. By engineering DNA constructs with hypoxia-responsive promoters, prodrug metabolizing enzymes may be selectively expressed at the tumour site (Dachs et al. 1997). However, delivery of vectors into solid tumours will be challenging. An alternative approach is to activate prodrugs using ionizing radiation. Radiolysis of water generates aquated electrons (eaq −), which may act as efficient reducing agents in lieu of endogenous enzymes. In addition, the radiation field may be focused onto the tumour to provide specificity in addition to hypoxia alone. This is a promising area of research to keep an eye on.
14.6.3 Patient Selection
Early clinical trials of tirapazamine – a hypoxia-activated prodrug – in advanced head and neck squamous cell carcinoma (HNSCC) demonstrated a significant decrease in locoregional failure versus 5-fluorouracil (5FU) combined with cisplatin and radiotherapy (Rischin et al. 2006). The subsequent phase III trial of tirapazamine, however, failed to meet its primary endpoints (Rischin et al. 2010). The key distinction between the two studies is that only the former used imaging to select for patients with evidence of tumour hypoxia. Recent meta-analyses of oxygen-electrode studies have further suggested that overall survival is compromised only in the subset of patients with extreme hypoxia (Nordsmark et al. 2005). Thus there is an urgent need to develop clinically applicable tools for the identification and quantification of tumour hypoxia.
Several imaging techniques are currently under development. In a study of 24 patients with prostate carcinoma, maps from blood oxygen level-dependent magnetic resonance imaging (BOLD MRI) yielded high sensitivity for defining hypoxic tumour regions stained with pimonidazole (Hoskin et al. 2007). However, it has limited specificity owing to influence from other factors including blood flow, CO2, hematocrit, and pH. Hypoxic regions may also be identified using positron emission tomography (PET) in combination with injectable tracers such as 18F-MISO (Rasey et al. 1996). However, the technical complexities and limited availability behind these methods would likely preclude a wider clinical application.
The ideal biomarker should be accessible through non-invasive methods, be specific to hypoxia, and be sensitive to changes in pathology upon disease progression or therapeutic response. One candidate biomarker is osteopontin (OPN) – high concentrations of plasma osteopontin (OPN) are associated with tumour hypoxia as measured by Eppendorf electrodes, as well as decreased overall survival across multiple tumour types (Weber et al. 2010). In support of this hypothesis, retrospective analysis of 320 patients with squamous cell carcinoma of the head and neck from the DAHANCA 5 trial demonstrated that patients with high pre-treatment OPN levels benefited from hypoxic modification with nimorazole, but those with low to intermediate levels did not (Overgaard et al. 2005). However, plasma OPN was not shown to confer adverse prognosis or predict response to tirapazamine in the large TROG 02.02 phase III trial. Thus the clinical significance of plasma OPN is unclear. Approaches combining multiple biomarkers have been met with greater success. In a study of 323 HNSCC patients, classification of tumours as hypoxic based on the expression of a 15-gene hypoxia signature independently predicted benefit from the hypoxic radiosensitizer nimorazole (Toustrup et al. 2012). Validation is currently on-going.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


