Fig. 15.1
Schematic showing the link between cellular stress and glycolysis. Stress stimuli trigger the stress signaling pathway such as p38/MAPK resulting in the activation of MAPK-M2 by phosphorylation. MAPK-M2 directly accelerates glycolysis by modulating PFKFB3 activity (indicated by brown arrows). Next, ROS, a major intracellular stress inducer also affects glycolysis by diverting glycolysis towards PPP (indicated by purple arrows). ROS affects the sulphydryl group of PKM2 resulting in its inactivation. Also, ROS promotes the stabilization of HIF1α which in turn activates several glycolytic enzymes resulting in an increase in the rate of glycolysis. Acidosis, another frequent and common intracellular stress, blocks LDH activity to redirect glucose oxidation via PPP (indicated by red arrows). Also, under acidosis p53 is up regulated which in turn promotes PPP by activating GPD. Enzymes are indicated in square boxes in bold and italicized letters. Grey arrows indicate the enzymes involved in glycolysis, black arrows indicate the sequential steps in glycolysis. Purple color refers to ROS mediated effects, red color refers to acidosis mediated effects and brown color refers to signaling mechanism mediated effects. MAPK Mitogen activated protein kinase, PFKFB3 -phosphofructokinase-2-kinase/fructose-2,6-bisphosphatase 3, HIF1α hypoxia inducible factor 1alpha (α), PPP pentose phosphate pathway, ROS reactive oxygen species, HKII hexokinase II, PGI phosphoglucose isomerase, GPD glucose -6-phosphate dehydrogenase, PFK phosphofructokinase, ALD aldolase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, TPI triose phosphate isomerase, PGK phosphoglycerate kinase, PGM phosphoglycerate mutase, ENO enolase, PKM2 pyruvate kinase M2 isoform, LDH lactate dehydrogenase, PPP pentose phosphate pathway, TCA cycle tricarboxylic acid cycle
15.3.1 ROS
It is widely known that excessive accumulation of ROS beyond cellular tolerance is cytotoxic. High rate of metabolism leads to an elevation in the level of cellular ROS creating an imbalance in the ratio of antioxidants and prooxidants. However, recent investigations have revealed that a minimal elevation in ROS prior to the chronic/injurious level could provide cue to cancer cells for the necessity of metabolic alteration. In order to escape ROS mediated injury and mitigate further cellular stress alternative pathways such as glycolysis and PPP are facilitated (Sosa et al. 2013).
One of the adaptive mechanisms recently identified is that during high levels of ROS, the enzyme, purvate kinase-M2 isoform (PKM2) is inactivated by the modification of its suphydryl group by ROS. PK-M2 catalysis the conversion of phosphoenol pyruvate into pyruvate for further oxidation into either acetyl coA or lactate. Thus inactivation of PK-M2 affects the glycolytic step of pyruvate formation diverting it towards the PPP (Anastasiou et al. 2011). The advantage of this altered metabolism is that PPP generates NADPH which can rejuvenate oxidized glutathione into its reduced form to act as an active antioxidant. This way, glucose is diverted away from mitochondrial oxidation which while reducing the level of ROS produced, simultaneously supports the replenishment of glutathione to neutralize the cellular ROS level (Dang 2012). It is noteworthy that the PKM2 has also been indicated as a gatekeeper of cell growth and survival (Harris et al. 2012).
An intracellular increase in ROS levels also has been known to stabilize HIF-1alpha, a key protein which transactivates several genes of glycolysis (Semenza et al. 1994). Among the glycolytic enzymes that are up regulated, the activation of PDK results in the rewiring of the metabolic circuitry of glucose catabolism. PDK phosphorylates PDH resulting in its inhibition, directing the pyruvate to be converted to lactate by LDH. Similarly, the activation of PFKFB4 results in the degradation of 2,6-FBP (an activator of PFK1 that catalyzes the conversion of fructose 1 phosphate into fructose 1,6 bisphosphate, a rate limiting step of glycolysis) (Yalcin et al. 2009). Such inhibition of PFKFB4 has been known to redirect glucose into PPP (Ros and Schulze 2013). However, PFKFB3 is also activated by HIF-1alpha, and PFKFB3 drives glucose into glycolysis. Depending upon the cellular requirement particular isoforms of PFKFB (3 or 4) can play a critical role in the adopting the mode of glucose catabolism. Thus ROS influences energy metabolism by facilitating glucose utilization by non-mitochondrial pathways (glycolysis/PPP) enabling cancer cells to evade chronic intracellular stress.
15.3.2 Acidosis
Cellular acidosis in general can be defined as a decrease in intracellular pH that can affect normal cell physiology, eventually causing cell death. Lactic acid (lactate) generated by glycolysis may contribute to intracellular acidosis yet it is not an indispensable factor. A change in the level of intracellular H+ concentration is sufficient to cause acidosis. In an elegant report, by experimental manipulation of intracellular lactate levels and intracellular H+ concentration, Jen-Tsan Chi’s group has demonstrated that acidosis can promote metabolic reprogramming (Lamonte et al. 2013). Under intracellular acidification, cancer cells favored the diversion of glycolysis into PPP (both oxidative and non-oxidative) and enhanced glutamine-metabolism to meet biosynthetic (PPP) and bioenergetic (glutaminolysis) demands. It is also evident that acidosis also governs the oxidative and nonoxidative PPP depending upon the cellular requirements. Under nonoxidative PPP conditions (R5P-pentose phosphate), any excess or accumulation could be redirected or reversed back to glycolysis by the reversible reactions of transketolase and transaldolase.
Elevated levels of free protons (H+) are often shuttled to the extracellular tumor microenvironment to maintain intracellular pH (pHi) at physiologic levels. Increasing amounts of H+ being pumped into the extracellular space creates an acidic microenvironment, which is known to select for cells with enhanced metastatic potential as well as provide resistance to chemotherapy (Bailey et al. 2012; Moellering et al. 2008; Schlappack et al. 1991). Thus cellular response to intracellular pH enables metabolic reprogramming to evade cellular acidification-related cytotoxicity.
15.3.3 Signal Transduction
Stress responsive signal transduction mechanisms have been known to play a significant role in the regulation of several processes including cell cycle checkpoints, apoptosis etc. Mitogen activated protein kinase (MAPK) pathways (e.g. p38) account for many of such changes in cellular processes. However, evidence for any direct regulation of glycolysis by a signal transduction molecule remained elusive. Recently it has been demonstrated that MAPK-M2 (MAPK-activated protein kinase 2) activates PFKFB3, a key promoter of glycolysis (Novellasdemunt et al. 2013). Thus metabolic reprogramming to glycolysis is achieved through signaling pathways as well. Similarly, STAT1-dependent expressional regulation of glycolysis suggests a potential role for STAT1 as a transcriptional modulator of genes responsible for glycolysis (Pitroda et al. 2009). Thus increasing data indicate that a coordinated network facilitates the metabolic switch from oxidative phosphorylation to glycolysis. Taken together intracellular stress inducers including ROS, pH and others could influence redirection of glucose metabolism away from mitochondria but towards glycolysis and PPP.
15.4 Targeting Glycolysis
15.4.1 Rationale
Apart from providing the energy source, the intermediates (substrates/products) of glucose metabolism (glycolysis) are used for anabolic reactions as well. For example, glucose-6-phosphate is used for synthesis of ribose -5 phosphate for further use in nucleic acid synthesis, similarly, dihydroxyacetone for lipid synthesis. Multiple lines of evidence show that the enhanced glucose uptake witnessed in tumor cells is to meet manifold requirements, and not just the energy demand. Thus, the enhanced glucose uptake is not just a favorite biochemical change, rather an indispensable metabolic transformation that is critical for the rapid, uncontrolled proliferation of tumor cells. In principle, targeting glucose metabolism essentially involves targeting more than one pathway that is interlinked with increased-glucose utilization.
Thus it is evident that aerobic glycolysis in conjunction with the PPP provide multiple benefits to cancer cells such as promoting tumor progression and providing resistance to therapy. Hence, this key signature of cancer cells, tumor metabolism, particularly the tumor glycolysis, provides an ideal target for therapeutic intervention.
Emerging data also substantiate several non-glycolytic functions of glycolytic enzymes and the metabolic intermediates of glycolysis (Fig. 15.2). Many enzymes of the glycolytic pathway such as hexokinase II (HKII), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase (PK)-M2 isoform and lactate dehydrogenase (LDH) participate in a number of subcellular functions including gene regulation and histone modifications (Kim and Dang 2005). Similar to the enzymes of glycolysis, some of the metabolic intermediates of glycolysis are also involved in non-glycolytic functions. Fructose-1, 6 bisphosphate by maintaining cytochrome C in an inactive state plays an anti-apoptotic role in cancer (Diaz-Ruiz et al. 2008). While pyruvate, another metabolic intermediate of glycolysis, is known to promote chemoresistance by the upregulation of p-glycoprotein (Wartenberg et al. 2010) its transporters (monocarboxylate transporters, MCTs) regulate CD147, a matrix metalloproteinase inducer (Izumi et al. 2011; Pertega-Gomes et al. 2011). Taken together, these findings demonstrate that glycolytic enzymes and metabolic intermediates play a key role beyond glycolysis, impacting cancer cell growth.
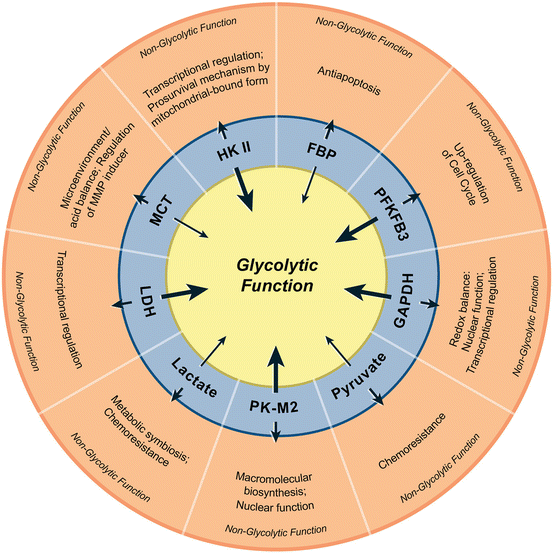
Fig. 15.2
Non-glycolytic functions of glycolytic enzymes and metabolic intermediates. In the innermost circle, thick arrows represent enzymes and thin arrows indicate intermediate metabolites. The short arrows pointing towards the outer circle represent the non-glycolytic functions of corresponding enzymes/metabolites. HKII hexokinase II, FBP fructose 1,6-bisphosphate, PFKFB3 6-phosphofructokinase-2-kinase/fructose-2,6-bisphosphatase 3, GAPDH glyceraldehyde-3-phosphate dehydrogenase, PKM2 pyruvate kinase M2, LDH lactate dehydrogenase, MCT monocarboxylate transporters (Reproduced with permission from Molecular Cancer (2013))
It is noteworthy that a higher lactate level has been known to correlate with aggressive phenotype including tumor recurrence and the metastatic potential resulting in poor prognosis (Walenta et al. 2000). Since elevated lactate levels indicate the preponderance of glycolysis, antiglycolytic agents could be very effective in targeting such metabolic-phenotype in tumors. It is increasingly evident that lactate export mitigates intracellular acidification while its import into normoxic cancer cells provides a substrate source for TCA cycle (mitochondrial oxidation) and energy production. Thus a “metabolic symbiosis” prevails within a tumor due to the metabolic heterogeneity, viz. a central hypoxic and glycolytic population of cells, and a peripheral oxygenated tumor cells (Sonveaux et al. 2008). The existence of such a depending upon tumor vasculature and “give and take lactate” mechanism (Semenza 2008) will benefit both lactate-exporting and lactate-importing cells.
15.4.2 Therapeutic Opportunities
Oncogenic driver mutations have been known to culminate in altered signal transduction pathways enabling tumor cells to reprogram their metabolic circuitry to adapt to the microenvironment. For example, it has been demonstrated that enhanced nutrient uptake is an effect of oncogenic RAS mutations (Yun et al. 2009). The tumor-specific shift in metabolism has been shown to be inevitable for uncontrolled proliferation and invasion of almost all solid tumors, hence the tumor metabolism is aptly described as “Cancer’s Achilles’ Heel” (Kroemer and Pouyssegur 2008). Thus, mounting evidence points to the notion that “enhanced or increased glucose uptake” of cancer cells could be a potential therapeutic target. Several reviews have emphasized and elegantly demonstrated the potential molecular targets of glycolysis that can be exploited for anticancer therapy. Figure 15.3 illustrates the biochemical steps that are blocked by currently explored inhibitors that are either under preclinical or clinical evaluation.
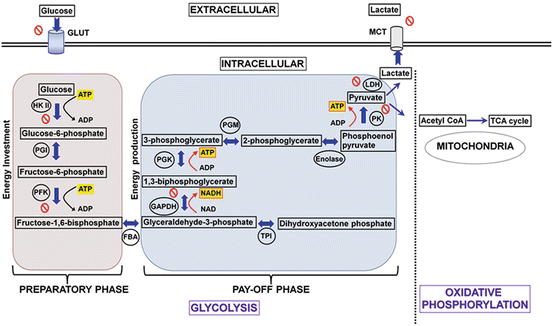
Fig. 15.3
Diagram showing the two phases of glycolysis and the molecular targets currently exploited for potential therapeutic drug strategies. Energy molecules such as ATP and NADH are highlighted in yellow, black arrows indicate consumption while red arrows indicate the energy release. The enzymes involved in respective reactions are abbreviated and encircled, where as the block symbol shows the targets exploited for drug development in preclinical investigations. NADH nicotinamide adenine dinucleotide reduced form, GLUT glucose transporters, HKII hexokinase II, PGI phosphoglucose isomerase, PFK phosphofructokinase, FBA fructose-bisphosphate aldolase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, TPI triose phosphate isomerase, PGK phosphoglycerate kinase, PGM phosphoglycerate mutase, PK pyruvate kinase, LDH lactate dehydrogenase, MCT monocarboxylate transporter (Reproduced with permission from Molecular Cancer (2013))
15.4.3 Targeting Tumor and the Microenvironment
Several lines of evidence indicate that the impact of metabolic reprogramming to glycolysis is not confined to cancer cells but extends to the stroma/fibroblasts in the tumor microenvironment which favors tumor progression by sustained fuel or energy supply. These microenvironment-glycolytic reprogramming are also orchestrated by oncogenes. It is important to recall that oncogenetic activation (RAS, NF-kB etc.) and tumor suppressor loss have also been shown to facilitate the metabolic reprogramming of tumor microenvironment. Thus, the cancer cell’s metabolic reprogramming to glycolysis also directs the metabolic reprogramming of cancer associated fibroblasts (Lisanti et al. 2013). It has been known that tumor microenvironment acts as a barrier and renders defense against therapeutic agents. Targeting tumor glycolysis will therefore affect the tumor microenvironment which could disrupt microenvironment-related protumorigenic properties.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


