Fig. 19.1
EMT induction. (a) Several cytokines, ROS, and hypoxic environment induce EMT through transcriptional factors, slug, ZEB1, twist, and others. (b) Lung adenocarcinoma cell line (A549) is induced EMT by TGF-β treatment (10 ng/mL) for 48 h
19.2.3 EMT in Lung Cancer
As similarly to other cancers, it had been also indicated the EMT in lung cancer in vitro and in vivo. In vitro, lung cancer cells altered to EMT cells in some lung cancer cells by means of TGF-β [26], IL-6 [22], and HGF [27]. These EMT cells had high motile and invasive potential. Moreover, poorly prognostic clinicopathological features had been described in NSCLC with EMT features. For example, NSCLC with high expression of E-cadherin had longer overall survival and less metastasis. On the other hand, NSCLC with high expression of mesenchymal markers and transcriptional factors to induce EMT had shorter overall survival and poor differentiation [8].
19.2.4 EMT-Induced Drug Resistance
In addition, it had been also suggested that EMT-induced lung cancer cells acquire the resistance for chemotherapy without any acquired gene mutation. The profile of less expression of E-cadherin and high expression of N-cadherin was indicated in patients with resistance to chemoradiotherapy including cisplatin in NSCLC [28]. The knockdown of snail inhibited the induction of EMT and its drug resistance to cisplatin in NSCLC cells [29]. These findings strongly support the ability of EMT to induce resistance to cytotoxic drugs in NSCLC. In addition, resistance to EGFR-TKIs is also demonstrated by accumulating evidences [5]. Transcriptional factors including ZEB1 and slug might play a crucial role in these resistances. TGF-β- and EGF-induced EMT acquired resistance to EGFR-TKIs through mTOR and MEK/Erk pathways in vitro [27, 30]; HGF-induced EMT cells acquired drug resistance for gefitinib in NSCLC cells and etoposide in small cell lung cancer (SCLC) cells [21, 31]. IL-6-induced EMT cells acquired drug resistance for gefitinib in NSCLC cells. Shien et al. reported that chronic exposure of gefitinib enables to induce EMT and resistance to EGFR-TKIs without any known mutations [32]. Clinical observations also indicated that several NSCLC with resistance to EGFR-TKIs demonstrated the EMT features in patients with NSCLC harboring EGFR mutation [33] (Fig. 19.2).
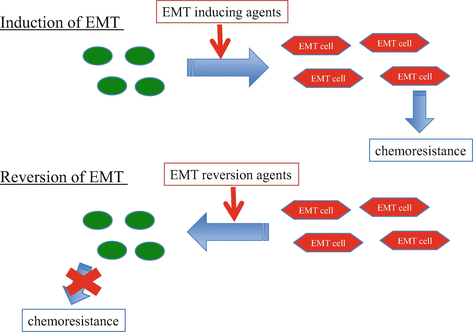
Fig. 19.2
Drug resistance in EMT. Induction of EMT promotes chemoresistance in lung cancer. Restoration of EMT also restores chemoresistance. Targeting EMT is a promising strategy to overcome drug resistance
19.2.5 Restoration of EMT-Induced Drug Resistance
The restorations of EMT-induced drug resistance were also reported. A MEK/Erk inhibitor and mTOR inhibitor suppressed EMT and improved drug sensitivity in lung cancer cells [27, 34–37]. It was reported that metformin, a widely used drug for diabetes, might decrease the frequency of several cancers [38, 39] and be associated with improved survival among patients with diabetes with stage IV NSCLC [38]. In vitro, it has been reported that metformin suppressed the proliferation of several cancer cells and reverse TGF-ß induced EMT in breast cancer cells [40–43]. In lung cancer, metformin significantly suppressed the regrowth of the tumors after withdrawing gefitinib treatment in xenograft mouse model of lung cancer cell in vivo [44]. Recently, Li L. et al. showed that metformin restored IL-6-induced EMT and drug resistance through inhibition of JAK/STAT3 pathways [22]. In addition, TTF-1 [45], crizotinib [31], which inhibits ALK and MEK in NSCLC, and the eukaryotic initiation factor inhibitor GC7 (N1-guanyl-1,7-diaminoheptane), [46] also decrease EMT-induced drug resistance in lung cancer cells. In other cancer, other drugs such as tranilast [47], resveratrol [48], propolis [49], and eribulin [50] have been also reported to affect EMT-induced drug resistance (Fig. 19.2).
19.3 Cancer Stem Cell
19.3.1 Characteristics of Cancer Stem Cell
The concept of CSC has been recently described as one of the mechanisms to establish the intratumoral heterogeneity [51]. Based on the hypothesis, the population of CSCs has the potential to self-renewal and to maintain cancer. Firstly, leukemia-initiating cells in acute myeloid leukemia were identified and these cells enabled to form tumor in SCID mice [9]. These cells had the profile of cellular surface antigen with positive for CD34 and negative for CD38 [52]. Afterward, solid tumor-initiating cells had been also isolated in succession. Cell surface markers vary from types of cancer. Breast cancer-initiating cells describe CD44+ and CD24−/low [10]. Brain tumor-initiating cells and colon cancer-initiating cells describe CD133+ [11, 53]. Moreover, other markers were also reported in several solid cancers. Any of these tumor-initiating cells enable to originate tumor, guide cells differentiation, and create the heterogeneity of tumor [51] (Fig. 19.3).
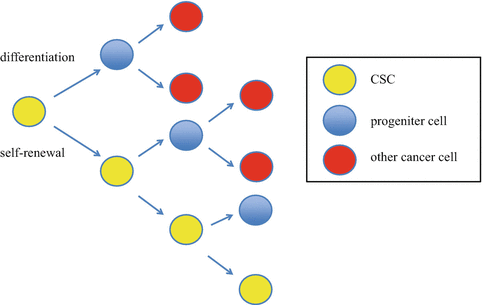
Fig. 19.3
Cancer stem cell model. Cancer stem cell is capable to self-renewal and differentiation. This characteristic causes initiating tumor and intratumoral heterogeneity
19.3.2 Association of EMT and CSC
It has been also described the association between EMT and CSC. In breast cancer cells, TGF-β-induced EMT cells included the cells with the stemness to create mammosphere forming and expression of the specific cell surface markers of CD44+ and CD24−/low [54]. CD44 involves in Wnt pathway, which plays an important role in EMT induction in breast cancer cells [55]. Cells with expression of CD133 and CXCR4, a chemokine receptor, had the high invasive potential in mesenchymal pancreas cancer cells [56]. Moreover, it had been also indicated that CSCs had drug resistance in several cancers, which was similar to EMT. These findings might suggest the potent association between EMT and CSCs.
19.3.3 CSC in Lung Cancer
In lung cancer cells, several reports suggested the presence of CSCs with cell surface markers of CD133, CD44, aldehyde dehydrogenase (ALDH), ABCB1, and CXCR4 [12, 57–59]. The profile of these markers still varies from reports. Moreover, it has been also suggested that lung cancer-initiating cells linked to EMT phenotype. Ectopic expressions of Oct4 and Nanog, which were some of transcriptional factors to make cells stemness in several cancers, induced high frequency of CD133-positive cells, sphere formation, drug resistance, and EMT with slug expression in lung cancer [60]. TGF-β treatment induced not only EMT but also sphere formation and expression of Oct4, Nanog, and CD133 in lung cancer cells [61].
19.3.4 Overcome CSC-Induced Drug Resistance
Recently, some treatment strategies for CSCs were indicated. Inhibition of TGF-β type I receptor with paclitaxel treatment improved the antitumor efficacy in CSC of breast cancer cells [62]. Inhibition of nodal/Activin receptor Alk4/Alk7 suppressed CSCs and increased drug sensitivity to gemcitabine in pancreas cancer [63]. BMP4 was one of the differentiation factors of colorectal cancer, and it suppressed the CSCs and improved drug sensitivity to 5-fluorouracil and oxaliplatin in colorectal cancer cells [64]. Histone deacetylates (HDAC) was also reported to differentiate cells to mesenchymal and induce CSCs [65, 66]. HDAC inhibitor was reported to suppress the CSCs in chronic myelogenous leukemia and some solid tumors in preclinical studies [67]. Notch signaling and cyclin-D1 pathway, which regulated cell cycle, induced EMT and are related to CSCs in breast cancer cells [68, 69]. Notch inhibitor was also studied. In NSCLC cells, it was reported that inhibition of Notch signaling [70, 71]; checkpoint protein kinase (Chk1) [72]; Bcl-XL, one of the anti-apoptosis factors [73]; all-trans retinoic acid [74]; and trifluoperazine, one of an antipsychotic agents [75], inhibited CSC growth and suppressed the acquisition of drug resistance in preclinical studies (Fig. 19.4).
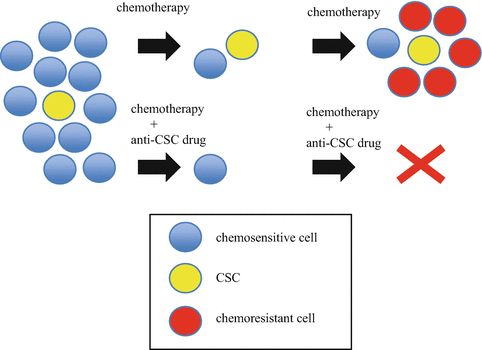
Fig. 19.4
Targeting cancer stem cell strategy. It has been thought that cancer stem cells (CSCs) have originally resistance to standard chemotherapies. Residual cancer cells after chemotherapy might induce acquired resistance to standard chemotherapies. Targeting CSC might overcome this process
In clinical, some drugs have been studied in a phase 1 or 2 clinical trial in patients with lung cancer (Table 19.1). The combination with romidepsin, a HDAC inhibitor, and erlotinib was reported in phase 1 clinical trial [76]. The combination of decitabine, a HDAC inhibitor, and valproic acid was also studied in phase 1 clinical trial for NSCLC [77]. Phase 1 clinical trial of demcizumab, which inhibits delta-like ligand 4 (DLL4) in the Notch signaling pathway, with carboplatin and pemetrexed for non-squamous NSCLC has been reported at ASCO 2015 Annual Meeting [78]. Phase 2 clinical trial of demcizumab has been initiated. Selumetinib, a MEK inhibitor, with docetaxel was studied for patients with NSCLC harboring KRAS mutation in phase 2 clinical trial [79, 80]. Selumetinib was also reported for maintenance therapy after platinum doublet therapy for patients with NSCLC at ASCO 2015 Annual Meeting [81]. Phase 1 clinical trial of tarextumab, a Notch inhibitor, was presented in patients with SCLC at ASCO 2015 Annual Meeting [81]. Phase 2 clinical trial of tarextumab has been initiated. Moreover, several other drugs were also studied in phase 1 trial for solid tumors.
Table 19.1
Already reported clinical trials of anti-CSC drug for lung cancer
Drug | Target | Histology | Combination | Clinical trial | References |
|---|---|---|---|---|---|
Romidepsin | HDAC | NSCLC | Erlotinib | 1 | [76] |
Decitabine | HDAC | NSCLC | Valproic acid | 1 | [77] |
Demcizumab | DLL4 (notch) | non-Sq NSCLC | CBDCA+PEM | 1 | [78] |
Selumetinib | MEK | NSCLC with KRAS mutation | DOC | 2 | |
Tarextumab | Notch | SCLC | CDDP+ETP | 1 | [81] |
19.4 Immuno-protective in EMT and CSCs
The immune-check point therapy is noteworthy treatment in several cancers. Programmed death -1 (PD-1) receptor is one of the targets of these treatments [82]. PD-1 receptor and PD-L1 expression play the important role for immune escape mechanism [82]. However, the characteristics of tumor cells with sensitivity for immune-check point therapy are still unknown. PD-L1 expression is now being watched with interest. Recently, a link between the induction of EMT and the overexpression of PD-L1 was reported. Chen L. et al. reported that ZEB1 promoted metastasis and increased expression of PD-L1 through inhibition of microRNA-200 in lung cancer cells [83]. Alsuliman A. et al. indicated that PD-L1 expression increased in TGF-ß-driven EMT cell and decreased by inhibition of PI3K or Erk in breast cancer cells [84]. In lung cancer, Ota K. et al. indicated that PI3K/Akt and MEK/Erk pathways mediate PD-L1 expression in NSCLC cells with either ALK-translocation or EGFR mutation [85]. Recently, we also demonstrated a close relationship between PD-L1 expression and EMT induction/reversion by several drugs [86]. These findings might suggest the possibility of EMT-related immune suppression and modification of immune-check point therapy. Further investing is needed.
19.5 Conclusion
EMT and CSCs were indicated as the key phenomena of acquisition or potentially resistance to chemotherapies. Therefore, the new strategies of combination with anti-EMT/CSCs treatment with chemotherapy might reduce the residual cells after chemotherapy and overcome the acquisition of resistance. Driving cancer cells into extinction is expected by further investigations.
References
1.
2.
3.
4.
Sharma SV, Lee DY, Li B et al (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141:69–80CrossRefPubMedPubMedCentral
5.
6.
Kajiyama H, Shibata K, Terauchi M et al (2007) Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 31:277–283PubMed
7.
8.
9.
10.
Al-Hajj M, Wicha MS, Benito-Hernandez A et al (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988CrossRefPubMedPubMedCentral
Related posts:
 Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
 Minor-Driver Mutant
Minor-Driver Mutant
 Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
 EGFR Mutant
EGFR Mutant
 PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


