Melanoma is the most aggressive form of skin cancer. Unfortunately, despite recent improvements for some solid tumors, the prevalence and mortality of melanoma continue to increase. The identification of activating mutations in melanoma, combined with a growing appreciation of the different pattern of genetic changes in the anatomically defined melanoma subtypes, has become the focus of a concerted effort to translate these discoveries into personalized therapeutic approaches for this disease. This article reviews the known mutations, amplifications, and deletions in kinase signaling pathways that have been implicated in melanoma; the prevalence of these genetic events in clinicopathologically defined melanoma subtypes; and the results of clinical trials that use targeted therapy approaches to block aberrantly activated pathways resulting from these mutations. The challenges that must be overcome to achieve improved outcomes with targeted therapies in melanoma in the future are also discussed.
Melanoma is the most aggressive form of skin cancer. Unfortunately, despite recent improvements for some solid tumors, the prevalence and mortality of melanoma continue to increase. For patients with distant metastases, treatment with single-agent or combination chemotherapy regimens have generally resulted in very low response rates, with no significant impact on patient survival. Immunotherapies (eg, interleukin-2, ipilimumab) have also yielded overall low response rates, although a small subset of patients have achieved durable responses and long-term survival. These modest achievements are further limited by noting that immunotherapies may also result in significant toxicities, including treatment-related deaths. Thus, a critical need exists for new therapeutic approaches for this aggressive disease.
The treatment of many cancers is entering a new era based on an improved understanding of the molecular pathogenesis of these diseases. Although some cancers seem to be primarily driven by viral infection, most are caused by genetic events that alter the expression or function of normal genes and proteins. These events, which include gene amplifications, deletions, and mutations, disrupt the regulatory processes that normally control the growth and survival of cells. Multiple analyses have shown that although a spectrum of genetic abnormalities is present in cancer cells, most affect certain signaling pathways and functions. Cancer cells are subsequently often critically dependent on these pathways for survival, a phenomenon termed oncogene addiction . This reliance on pathways that are hyperactivated by genetic events occurring specifically in cancer cells presents a therapeutic opportunity to block those targets to inhibit the growth and survival of the cancer while sparing the normal cells of the body. This approach, termed targeted therapy , has been shown to be effective and is approved by the U.S. Food and Drug Administration (FDA) for treating several cancers, including chronic myelogenous leukemia, gastrointestinal stromal tumors (GISTs), and renal cell carcinoma. Targeted therapies have also been effective in treating specific subpopulations of patients with other cancers, such as trastuzumab (Herceptin) for treating HER2/neu-amplified breast cancer. For each of these examples, successful implementation of a targeted therapeutic approach was critically dependent on the identification of activating genetic events and the affected pathways that were present for each specific tumor type.
Evidence now shows that most melanomas harbor one or more mutations in critical kinase signaling pathways. Accumulating data support that the prevalence of these events varies greatly among the melanoma subtypes that have been defined by clinical and pathologic characteristics. Most cutaneous melanomas (CMs) arise from melanocytes on sun-exposed skin. Exposure to ultraviolet radiation is thought to play a major causative role in these tumors. However, the role of ultraviolet radiation is less clear for cutaneous melanomas arising from relatively sun-protected sites. Examples of these melanomas include those arising on the palms and soles, termed acral lentiginous melanomas (ALMs). Melanomas may also arise from melanocytes in the mucosa of the head and neck, the gastrointestinal tract, and the genitourinary tract. Melanomas arising in these sites are classified as mucosal melanomas , and clearly arise in the absence of exposure to ultraviolet radiation. Consistent with the hypothesis that these melanoma subtypes are caused by different factors, comparative genomic hybridization (CGH) analysis has shown that these clinically defined groups of tumors have markedly different patterns of DNA copy number changes, including subtype-specific gene amplifications and deletions. CMs that arise in areas with chronic sun exposure and that have histologic evidence of chronic sun damage (CSD) also exhibit markedly different chromosomal and gene copy number changes compared with cutaneous melanomas without chronic sun damage. Melanomas may also arise from melanocytes in the uveal tract of the eye (iris, ciliary body, and choroid) and are referred to as uveal melanomas . These tumors are also characterized by chromosomal changes that are distinct from cutaneous, acral lentiginous, and mucosal melanomas.
The identification of activating mutations in melanoma, combined with a growing appreciation of the different pattern of genetic changes in the anatomically defined melanoma subtypes, has become the focus of a concerted effort to translate these discoveries into personalized therapeutic approaches for this disease. This article reviews the known mutations, amplifications, and deletions in kinase signaling pathways that have been implicated in melanoma; the prevalence of these genetic events in clinicopathologically defined melanoma subtypes; and the results of clinical trials that use targeted therapy approaches to block aberrantly activated pathways resulting from these mutations. The challenges that must be overcome to achieve improved outcomes with targeted therapies in melanoma in the future are also discussed.
BRAF
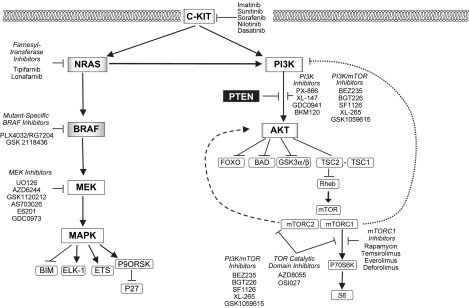
The RAS-RAF-MEK-MAPK signaling pathway is a critical regulator of cellular growth and survival ( Fig. 1 ). The first components of the pathway are the RAS-family GTP-ases. The RAS family members (HRAS, KRAS, NRAS) are guanine-nucleotide binding proteins that are embedded in the inner surface of the cell membrane. Normally, the RAS proteins are GDP-bound and inactive. Various activating signals result in the exchange of GTP for GDP, which activates RAS. RAS family members are also frequently affected by mutations in cancer that result in constitutive GTP-binding. The activated RAS members physically interact with the RAF family of serine-threonine protein kinases (ARAF, BRAF, CRAF) downstream of RAS. This interaction activates the RAF proteins, which then translocate to the cytoplasm and phosphorylate the MEK protein kinases (MEK1, MEK2). The MEK proteins are activated by this phosphorylation, and subsequently phosphorylate the P44/42 MAPK serine-threonine kinases (ERK1, ERK2). The activated MAPKs phosphorylate various transcription factors and cytosolic proteins to promote proliferation and survival.
In 2002, an experimental screen for mutations in RAF family members in cancer cell lines and tumors identified point mutations in BRAF in approximately half of the melanomas that were examined in the study, and occasional (3%–18%) colon, lung, breast, and ovarian cancer specimens. Since this sentinel observation, the high frequency of point mutations in BRAF in melanoma has been confirmed in multiple studies. A recent meta-analysis of more than 200 published studies reported an overall mutation rate of 65% in melanoma cell lines and 42% in tumors. The higher frequency of mutations in cell lines likely reflects a positive selection for cells with the BRAF mutation to propagate in vitro. Analysis of anatomic subtypes showed that BRAF mutations are common in cutaneous (42.5%) but uncommon in mucosal melanomas (5.6%) and rare in uveal melanomas (<1%) ( Table 1 ). Among cutaneous melanomas, BRAF mutations are common in superficial spreading melanomas (53%) but less prevalent in acral lentiginous (18%) and lentigo maligna melanomas (9%). Lentigo maligna melanomas are associated with CSD, and often originate in the head and neck region. The low prevalence of BRAF mutations in these tumors is consistent with the different patterns of DNA copy number gains and losses observed in the CGH analysis of cutaneous melanomas with or without CSD. BRAF mutations are also detectable in up to 80% of common acquired nevi. However, BRAF mutation rates are lower in several of the less-common nevi types, including congenital, Spitz, and blue nevi.
| Melanoma Subtype | BRAF Mutation | NRAS Mutation | c-KIT Mutation | c-KIT Amplification | GNαQ Mutation |
|---|---|---|---|---|---|
| Sun-exposed cutaneous | 43% | 26% | Non-CSD: <2% CSD: 2%–17% | Non-CSD: 0%–7% CSD: 6% | <1% |
| Acral | 18% | 4% | 18% | 24% | <1% |
| Mucosal | 6% | 14% | 24% | 26% | <1% |
| Uveal | <1% | <1% | <1% | <1% | 45% |
Approximately 50 different point mutations in BRAF have been identified in cancer. A single substitution, the BRAF V600E mutation, comprises approximately 85% of the BRAF mutations detected in melanoma. The V600E mutation increases the in vitro kinase activity of the BRAF protein more than 400-fold. Most of the other reported somatic BRAF mutations, particularly other changes involving the V600 residue, also increase BRAF’s catalytic activity (5- to 700-fold). A few BRAF mutations that have been detected in cancer cells ( G466E , G466V , G596R , D594V ) decrease the catalytic activity of the BRAF protein. Because BRAF V600E is the predominant mutation identified in tumors, including melanoma, most functional studies have examined the function of the protein encoded by this change. Expression of the BRAF V600E protein results in constitutive phosphorylation and activation of MEK and MAPK. Inhibition of BRAF V600E with small interfering RNA (siRNA, shRNA) inhibits MAPK activation, growth, and survival of human melanoma cell lines with this mutation. These data support that melanomas with the BRAF V600E mutation depend on it for survival, and thus implicated this mutation as a therapeutic target.
Sorafenib was the first BRAF inhibitor to be used in clinical trials in melanoma. Sorafenib is a small molecule inhibitor of multiple tyrosine kinases, including BRAF, CRAF, c-KIT, vascular endothelial growth factor receptor (VEGFR), and platelet-derived growth factor receptor (PDGFR). In preclinical studies, sorafenib slowed the growth of melanoma xenografts in nude mice, but it did not cause tumor regression. Among 34 evaluable patients with metastatic melanoma in a phase II trial, only one partial response was observed. Subsequently, more promising results were reported in a phase I/II clinical trial of sorafenib combined with paclitaxel and carboplatin. Although the trial enrolled patients with multiple tumor types, all of the clinical responses were achieved in patients with melanoma; among these patients, the response rate was 42% and the median progression-free survival was approximately 10 months. These results were encouraging when compared with previous trials in melanoma with paclitaxel and carboplatin alone (response rates of <10%–20%) ; however, a subsequent randomized phase III trial showed that sorafenib did not improve the response rate or progression-free survival compared with the doublet of paclitaxel and carboplatin alone.
These results raised the possibility that mutant BRAF was not a good therapeutic target in melanoma. The observation that activating BRAF mutations are present in up to 80% of benign nevi—indolent lesions with almost no malignant potential—is certainly consistent with the hypothesis that this genetic alteration alone cannot fully explain the aggressive biology of melanoma. Similarly, introduction of the BRAF V600E mutation alone was not sufficient to transform melanocytes into invasive lesions in multiple models, but rather required complementation by other genetic events to transform cells. An alternative explanation for the failure of sorafenib is that mutant BRAF is actually a good therapeutic target, but that the drug did not inhibit BRAF effectively. Sorafenib has shown marked clinical efficacy in renal cell carcinoma, a tumor characterized by dependence on VEGFR signaling but not BRAF. In the phase II trial of paclitaxel, carboplatin, and sorafenib, no association was noted between the presence of BRAF mutations and clinical responses, but a positive association was seen with expression of the VEGFR2 protein and clinical response.
The potential of the BRAF V600E protein as a therapeutic target for melanoma has now been validated by clinical trials with second-generation BRAF inhibitors. PLX4032 (also known as RG7204) is a more potent BRAF inhibitor than sorafenib, and it is selective for the V600E mutant form of the protein. PLX4032 inhibits the catalytic activity of the BRAF V600E protein at an IC50 of 13 nM, which is more than 10-fold lower than the dose that inhibits the wild-type protein. In contrast to sorafenib, an inhibitor of multiple protein kinases at therapeutic drug levels, PLX4032 has minimal activity against most other protein kinases, with an IC50 greater than 1000 nM for many related proteins. Preclinical studies showed that PLX4032 inhibited the growth of melanoma cells with a BRAF mutation at 10 to 100 times lower concentrations than melanoma cell lines without a mutant BRAF . PLX4032 also caused the regression of BRAF -mutant melanoma xenografts in mouse models.
Recently, the results of a phase I clinical trial with PLX4032 were reported. In the initial phase of the trial, the drug was well tolerated but no clinical responses were observed. However, the serum levels achieved were below those that correlated with antitumor activity in vitro. The drug was then reformulated from a crystalline compound to a microprecipitated bulk powder. Subsequently, linear dose-dependent increases in serum levels were observed, as were clinical responses. In the phase I trial dose expansion cohort of 32 patients with BRAF V600E –mutant melanoma treated with 960 mg twice daily (the dose selected for further testing), 2 complete and 24 partial responses were observed, for an overall response rate of 81%. The major toxicity was the development of cutaneous squamous cell carcinomas, mostly keratoacanthomas, which developed in 31% of patients. These lesions were treated with surgical resection and did not result in any patient coming off study. No squamous cell carcinomas were observed at noncutaneous sites. Although this high overall response rate is unprecedented for a single agent in melanoma, several patients developed secondary resistance after their initial response, noted by tumor recurrence or progression. The median duration of response to PLX4032 in the initial trial report was approximately 7 months.
The efficacy of targeting mutant BRAF is also supported by early results from the phase I trial of GSK2118436, another potent, mutant-specific BRAF inhibitor. Although the maximum tolerated dose has yet to be reached, patients with BRAF -mutant melanoma treated with 150 to 200 mg twice daily had a 63% response rate, and 39% of patients treated with lower doses had also experienced a response. The duration of these responses is currently unknown. Similar to the PLX4032 trial, cutaneous squamous cell carcinomas were the major toxicity observed in the GSK2118436 trial.
The reported activities of PLX4032 and GSK2118436 in patients with BRAF V600E –mutant melanoma suggest that a new standard of care likely will soon exist for these patients. However, it is also apparent that these agents should not be used in unselected patients with melanoma. In the phase I trial of PLX4032, five patients were included who did not have a BRAF mutation. None of these patients experienced a response; in fact, four experienced tumor progression in the first 2 months of treatment. The lack of clinical response, and possible rapid progression, in patients not harboring a BRAF mutation is consistent with observations in preclinical models by four different groups, suggesting potential promotion of tumor growth when mutant-selective BRAF inhibitors are used to treat BRAF wild-type melanomas. In these experiments, inhibition of BRAF in melanoma cell lines expressing wild-type BRAF resulted in the hyperactivation of the MAPK pathway. Although the results varied somewhat among the groups, the BRAF inhibitors promoted the formation of heterodimers of BRAF and CRAF that potently activate MEK in cells without a BRAF mutation. The cells were then dependent on CRAF for MAPK pathway activation and survival. Low–catalytic-activity BRAF mutants seem to activate MAPK similarly, and preclinical evidence suggests that melanoma cells with these mutations are sensitive to CRAF inhibition, including treatment with sorafenib. Thus, although sorafenib failed to show activity in unselected patients, it may be effective for certain genetic subtypes of melanoma.
NRAS
Mutations in the members of the RAS family of GTP-ases are one of the most frequent events in cancer. Although mutations of HRAS and KRAS are common in many cancer types, they are rare in melanoma. However, NRAS mutations have been reported in 14% of human melanoma cell lines and 15% to 25% of melanoma clinical specimens. The mutations affecting NRAS are highly conserved; mutations affecting codons 12, 13, and 61 constitute approximately 90% of the mutations reported in melanoma. The prevalence of NRAS mutations varies across the different anatomically defined melanoma subgroups, although not as dramatically as observed with BRAF mutations (see Table 1 ). NRAS mutations are detected in approximately 26% of cutaneous, 14% of mucosal, and fewer than 1% of uveal melanomas. Among cutaneous melanomas, 22% of superficial spreading melanomas and 28% of nodular melanomas have NRAS mutations, whereas significantly lower rates are observed in acral lentiginous (4%), spitzoid (10%), and lentigo maligna melanomas (0%). NRAS mutations are also present in common acquired nevi (6%–20%) at a similar rate as has been detected in melanomas, and potentially at an even higher rate in congenital nevi. Although rare in melanoma and other types of melanocytic nevi, HRAS mutations have been reported in 12 to 29% of Spitz nevi.
With rare exceptions, the common activating BRAF and NRAS mutations are mutually exclusive in melanoma tumor and cell lines, probably because both of these mutations potentially activate the MAPK pathway, and the presence of both would be functionally redundant. In contrast, approximately 10% of melanomas with BRAF mutations that are catalytically inactive (ie, D594V) also have activating NRAS mutations. Although the activated mutant forms of BRAF and NRAS both activate MEK and MAPK, the activation of these downstream elements is CRAF-dependent in melanomas with NRAS mutations, whereas it is BRAF-dependent in BRAF- mutant cells.
The development of direct RAS inhibitors, a priority in several cancer types, has been challenging. One approach has involved the development of farnesyl transferase inhibitors (FTIs), because RAS proteins must be farnesylated to translocate to the plasma membrane and activate their related signaling pathways. However, when farnesylation is inhibited, both NRAS and KRAS undergo an alternative modification, geranylgeranylation, that allows the proteins to be recruited to the plasma membrane. In addition, because more than 60 cellular proteins have been shown to be farnesylated, FTIs will likely have off-target effects (and potentially dose-limiting toxicities) that compromise the ability to reach levels that effectively inhibit RAS. This lack of specificity also makes the role of RAS in both the activity and toxicity of FTIs challenging to determine.
An alternative strategy to treat NRAS -mutant tumors is to inhibit pathways that are downstream of the mutant RAS protein. Experiments in multiple tumor types have shown that mutant RAS activates multiple prosurvival and proliferative pathways in addition to the RAF-MEK-MAPK cascade. Other RAS effectors include PI3K, RALGDS, and PKCϵ. Among these, PI3K has gained particular attention, because the PI3K-AKT pathway has been implicated in melanoma by other genetic events, and is the target of aggressive drug development (see Fig. 1 ).
NRAS
Mutations in the members of the RAS family of GTP-ases are one of the most frequent events in cancer. Although mutations of HRAS and KRAS are common in many cancer types, they are rare in melanoma. However, NRAS mutations have been reported in 14% of human melanoma cell lines and 15% to 25% of melanoma clinical specimens. The mutations affecting NRAS are highly conserved; mutations affecting codons 12, 13, and 61 constitute approximately 90% of the mutations reported in melanoma. The prevalence of NRAS mutations varies across the different anatomically defined melanoma subgroups, although not as dramatically as observed with BRAF mutations (see Table 1 ). NRAS mutations are detected in approximately 26% of cutaneous, 14% of mucosal, and fewer than 1% of uveal melanomas. Among cutaneous melanomas, 22% of superficial spreading melanomas and 28% of nodular melanomas have NRAS mutations, whereas significantly lower rates are observed in acral lentiginous (4%), spitzoid (10%), and lentigo maligna melanomas (0%). NRAS mutations are also present in common acquired nevi (6%–20%) at a similar rate as has been detected in melanomas, and potentially at an even higher rate in congenital nevi. Although rare in melanoma and other types of melanocytic nevi, HRAS mutations have been reported in 12 to 29% of Spitz nevi.
With rare exceptions, the common activating BRAF and NRAS mutations are mutually exclusive in melanoma tumor and cell lines, probably because both of these mutations potentially activate the MAPK pathway, and the presence of both would be functionally redundant. In contrast, approximately 10% of melanomas with BRAF mutations that are catalytically inactive (ie, D594V) also have activating NRAS mutations. Although the activated mutant forms of BRAF and NRAS both activate MEK and MAPK, the activation of these downstream elements is CRAF-dependent in melanomas with NRAS mutations, whereas it is BRAF-dependent in BRAF- mutant cells.
The development of direct RAS inhibitors, a priority in several cancer types, has been challenging. One approach has involved the development of farnesyl transferase inhibitors (FTIs), because RAS proteins must be farnesylated to translocate to the plasma membrane and activate their related signaling pathways. However, when farnesylation is inhibited, both NRAS and KRAS undergo an alternative modification, geranylgeranylation, that allows the proteins to be recruited to the plasma membrane. In addition, because more than 60 cellular proteins have been shown to be farnesylated, FTIs will likely have off-target effects (and potentially dose-limiting toxicities) that compromise the ability to reach levels that effectively inhibit RAS. This lack of specificity also makes the role of RAS in both the activity and toxicity of FTIs challenging to determine.
An alternative strategy to treat NRAS -mutant tumors is to inhibit pathways that are downstream of the mutant RAS protein. Experiments in multiple tumor types have shown that mutant RAS activates multiple prosurvival and proliferative pathways in addition to the RAF-MEK-MAPK cascade. Other RAS effectors include PI3K, RALGDS, and PKCϵ. Among these, PI3K has gained particular attention, because the PI3K-AKT pathway has been implicated in melanoma by other genetic events, and is the target of aggressive drug development (see Fig. 1 ).
Related posts:
 Melanoma Pathology: Important Issues for Clinicians Involved in the Multidisciplinary Care of Melanoma Patients
Melanoma Pathology: Important Issues for Clinicians Involved in the Multidisciplinary Care of Melanoma Patients
 Surgical Approach to Primary Cutaneous Melanoma
Surgical Approach to Primary Cutaneous Melanoma
 Sentinel Lymph Node Biopsy for Melanoma: Critical Assessment at its Twentieth Anniversary
Sentinel Lymph Node Biopsy for Melanoma: Critical Assessment at its Twentieth Anniversary
 Immunotherapy of Melanoma: An Update
Evidence-Based Follow-up for the Patient with Melanoma
Immunotherapy of Melanoma: An Update
Evidence-Based Follow-up for the Patient with Melanoma
 Assessment of Patient-Reported Outcomes in Patients with Melanoma
Assessment of Patient-Reported Outcomes in Patients with Melanoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


