Outline
Foundation of Targeted Therapy
Vascular Endothelial Growth Factors and Vascular Endothelial Growth Factor Receptors
Agents Targeting the Vascular Endothelial Growth Factor Pathway
Agents Targeting Vascular Endothelial Growth Factor Receptors
Agents Targeting Multiple Vascular Endothelial Growth Factor–Related Molecules
Phosphatidylinositol-3-Kinase/AKT Pathway
Poly-ADP-Ribose Polymerase Pathway
Epidermal Growth Factor Receptor Pathway
Small Molecule Inhibitors Targeting Epidermal Growth Factor Receptor
Monoclonal Antibodies Targeting Epidermal Growth Factor Receptor
Unique Toxicities of Targeted Therapy
Special Considerations for Targeted Therapies
Key Points
- 1.
The development of new and effective therapies should be rooted in a clear understanding of tumor biology.
- 2.
Bevacizumab, a humanized monoclonal antibody to human vascular endothelial growth factor, was the first US Food and Drug Administration–approved drug targeting angiogenesis
- 3.
Reverse protein leukoencephalopathy is a rare complication of antiangiogenic agents, including bevacizumab, sorafenib, and sunitinib.
- 4.
As knowledge regarding tumor biology and pathogenesis continues to expand, the number of targeted agents will no doubt increase exponentially. These agents have the potential to have a profound impact on the treatment of patients with gynecologic malignancies.
Targeted Therapy
The use of site-specific combinations of surgery, chemotherapy, and radiation therapy in gynecologic malignancies has led to marked initial improvements in patient survival. Unfortunately, over the past 30 years, there has been little improvement in disease-specific mortality rates from the three major gynecologic malignancies ( Table 18.1 ). Furthermore, the incidence of these cancers has remained fairly stable since 1985, especially among endometrial cancers ( Table 18.2 ). The addition of traditional cytotoxic chemotherapies to current treatment regimens is likely to offer only increased toxicities in the absence of further survival benefit. To critically affect patient outcome and improve quality of life, the therapeutic armamentarium of modern oncologists must be expanded. The search for novel therapeutic options has led to an exploration of therapies targeting molecular pathways critical to the survival of cancer cells.
| 1975 | 1985 | 1995 | 2005 | 2008–2012 | |
|---|---|---|---|---|---|
| Cervical | 5.55 | 3.82 | 3.24 | 2.42 | 2.3 |
| Endometrial | 5.28 | 4.61 | 4.15 | 4.12 | 4.4 |
| Ovarian | 9.84 | 9.08 | 9.12 | 8.66 | 7.7 |
| 1975 | 1985 | 1995 | 2005 | 2008–2012 | |
|---|---|---|---|---|---|
| Cervical | 14.79 | 10.23 | 8.91 | 6.86 | 7.7 |
| Endometrial | 35.47 | 25.23 | 25.33 | 24.97 | 25.1 |
| Ovarian | 16.32 | 16.55 | 14.59 | 12.96 | 12.1 |
Foundation of Targeted Therapy
The development of new and effective therapies should be rooted in a clear understanding of tumor biology. In a pair of seminal papers in 2000 and 2011, Hanahan and Weinberg described the key capabilities acquired by normal cells that lead to the development of cancer. Fig. 18.1 represents the 10 “hallmarks of cancer” implicated in the tumorigenic process. Sustained proliferative signaling and evading growth suppressors relate to the development of cellular autonomy, which is essential for uncontrolled proliferation. Enabling replicative immortality indicates that the cell is unencumbered by two typical processes of senescence: (1) the cessation of growth upon reaching a set number of cellular doublings and (2) crisis, which involves massive cellular death. Resisting cell death is a feature seen in most cancer types, allowing the cells to continue to grow and replicate in the setting of damage that would lead to attrition in a normal cell. The ability to activate invasion of tissues and metastasis is crucial to the continued expansion of tumor when space and nutrients become limited. Inducing sustained angiogenesis describes uncontrolled growth of new blood vessels, which supply oxygen and nutrients to the tumor. Evading immune destruction allows the tumor to avoid detection by immune cells and limit the extent of immunologic killing. Finally, the ability to reprogram energy metabolism allows adjustment of energy use and creation to prioritize tumor growth and survival. Two additional enabling characteristics allow the cancer to leverage the above mechanisms into cellular growth and survival. Genomic instability hampers the cell’s ability to detect and repair DNA errors and damage. Thus, mutations accumulate, and genomic integrity is lost. The presence of rich immune infiltrates in the tumor microenvironment can support tumorigenesis by growth and prosurvival factors. Collectively, these steps enable tumor initiation, growth, and progression.
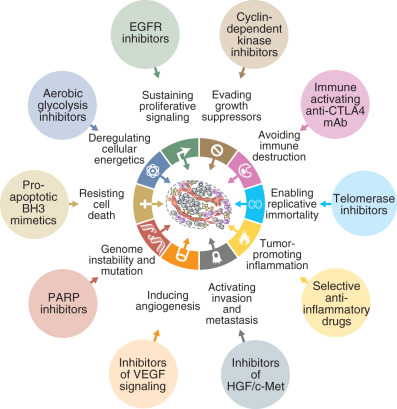
Acquisition of survival capabilities by cancer cells is theorized to be directly related to dysfunction of the normal molecular mechanisms and pathways within the cell and surrounding microenvironment. Because these pathways drive the progression of cancer, identifying and targeting the changes in the pathways to treat malignancy is a rational strategy. Thus, the field of targeted therapy has flourished in modern cancer treatment, especially among common gynecologic malignancies.
Cytotoxic chemotherapy typically acts primarily on any rapidly dividing cells. Although this may have the desired effects on tumor cells, these drugs do not discriminate between tumor cells and normal host cells, resulting in undesirable side effects in the gastrointestinal (GI) tract, bone marrow, and integumentary and other systems. Ideal targeted therapies provide a more directed approach by acting on targets selectively leveraged in tumor cells or in the tumor microenvironment. These targets are typically members of the pathways involved in tumorigenesis, supporting growth, proliferation, metastasis, and angiogenesis. By honing in on those pathways rather than broad-based activity, normal tissues could be spared, and adverse events are minimized.
These therapies hold the potential to reduce mortality rates from gynecologic malignancies while concurrently reducing the morbidity associated with cancer treatment by targeting abnormal rather than normal tissue. This chapter provides a broad overview of the pertinent molecular pathways in gynecologic cancer and the targeted agents that are currently being explored as treatment options. In addition, the unique toxicities of these targeted agents are reviewed. As our knowledge continues to expand, there will no doubt be myriad other pathways to exploit along with agents used to treat gynecologic malignancies.
Targeted Agents
Targeting molecular pathways that drive tumor progression can be accomplished through a variety of mechanisms. The first is a humanized monoclonal antibody (mAb), which is created to bind cancer-associated antigens or molecules for cancer therapy. mAbs may be directed toward ligands or cell surface molecules that participate in pathways of tumorigenesis. Table 18.3 describes the standard nomenclature of the mAb classes. These agents are classified based on the origin of the antibody. By definition, mAbs have affinity for a single target, which allows for minimal non–tumor-related effects. These agents are administered systemically and have long clearance times, allowing biweekly to monthly administration. One such agent is bevacizumab, which is an mAb to vascular endothelial growth factor A (VEGF-A), a key active ligand in angiogenesis, to be discussed later in the chapter. Engineered antibodies are also being developed that can interact with more than one ligand or binding domain.
| Suffix | Antibody Class | HAMA Potential | Example |
|---|---|---|---|
| “-omab” | Murine | +++ | |
| “-ximab” | Chimeric | +/++ | Cetuximab |
| “-zumab” | Humanized | + | Bevacizumab |
| “-mumab” | Fully human | − | Panitumumab |
Small molecule inhibitors are primarily oral agents that inhibit the function of molecular receptors through the blockage tyrosine kinase activity. Tyrosine kinases are enzymes involved in an array of normal and abnormal cellular functions. Their activity causes the transfer of a phosphate group from adenosine triphosphate (ATP) to a downstream protein tyrosine residue, resulting in changes in protein conformation and association, affecting innumerable biologic processes. Two types of tyrosine kinases exist: receptor and nonreceptor tyrosine kinases. Fig. 18.2 demonstrates the typical structure of a receptor tyrosine kinase, exhibiting three major domains: extracellular ligand binding, transmembrane, and cytoplasmic. Currently, approximately 100 receptor tyrosine kinases have been identified and classified. Nonreceptor tyrosine kinases are typically present in the cytoplasm or nucleus and interact with transmembrane receptors to phosphorylate downstream substrates. They may also be activated through signals derived from extracellular processes such as ion exchange. Erlotinib is one small molecule inhibitor that targets the epidermal growth factor receptor (EGFR) pathway affecting cellular division and proliferation.
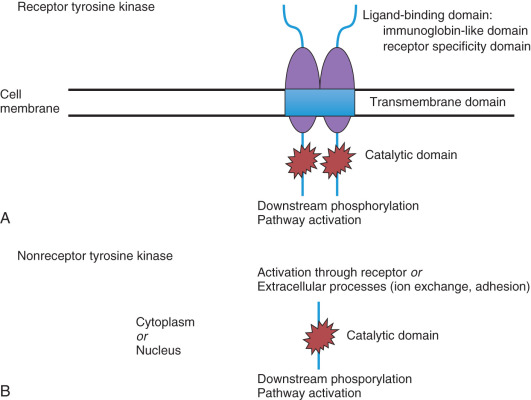
Small molecule inhibitors typically block tyrosine kinase phosphorylation through interaction with the ATP-binding site on the intracellular domain of the tyrosine kinase. Binding to this site may be reversible or irreversible, depending on the agent. These molecules often have short half-lives, which necessitates frequent administration. Secondary to the high degree of homology found at the ATP-binding domain of the various tyrosine kinases, many of these molecules may inhibit one or more receptors within the cellular mechanism. Depending on the pathways affected, this can improve antitumor activity or lead to undesired adverse effects related to “off-target” inhibition.
To reduce off-target associations, unique structural features of a given target are being identified. Then agents that are chemically altered to maximize interaction with these specific components can be introduced. One example is the development of wrapping technology. This involves targeting a defect that is inherently specific to a given region in a protein called a dehydron. Dehydrons are hydrogen bonds of the protein that are poorly protected from water. Drugs designed to protect a given dehydron from water will selectively direct ligands to the receptor with that specific defect. Additional methods of introducing selectivity are being explored, including the disruption of protein–protein interactions.
The use of RNA interference technology has great promise for expanding targeted therapy through knockdown of expression of specific genes involved in tumorigenesis. RNA silencing pathways are mechanisms within the cell that directly control gene expression. In general, short interfering RNAs (siRNAs) are generated from double-stranded RNA (dsRNA) within the cell by an endonuclease called Dicer. The antisense strands of the siRNA then associate with an RNA-induced silencing complex (RISC) that targets a specific messenger RNA (mRNA) for cleavage by Argonaute 2. Cleavage of the mRNA can silence genes involved in cellular survival, proliferation, invasion, and metastasis. Of note, the level of expression of the components of RNA silencing pathways correlates with survival in a variety of cancers.
The delivery of siRNA in vivo has been challenging. However, local delivery (intranasal or intravitreal) approaches have been advanced into clinical trials in nonmalignant diseases such as macular degeneration. Recent methods of delivery using biodegradable lipids and polymers have shown safety and efficacy in cancer models. Furthermore, an ongoing first-in-human phase I trial has demonstrated successful delivery of siRNA through nanoparticles. Early results from this trial revealed induction of RNA interference mechanisms at specific targeted sites. Nanodelivery vehicles for selective delivery can further enhance tumor specificity. This approach to targeted therapy has the potential to greatly expand the possible targets while reducing undesirable adverse effects.
Other unique agents have also been explored as prospective biologic options to disrupt carcinogenic pathways. Decoy receptors that bind key ligands of carcinogenic pathways have been developed. An example of this is VEGF-Trap (aflibercept), which binds VEGF to negatively affect angiogenesis. Antibody–drug conjugates (ADCs) combine a highly toxic cytotoxic agent with a specific immunoglobulin meant to create a targeted therapy that spares normal tissues that lack expression of the selected target. Current ADCs in development use targets including CD30, mesothelin, and folate receptor α. There is no doubt that as our knowledge of cellular signaling mechanisms grows, so will the variety of agents for targeted therapy.
Angiogenesis
Arguably, the most successful examples of targeted therapy in gynecologic cancer are found in the arena of angiogenesis. Angiogenesis is a key process for the supply of nutrients, oxygen, growth factors, and dissemination of a tumor. Thus, the development of new vasculature is an essential process for a tumor to grow beyond 1 mm in size. There are two primary mechanisms for the growth of new blood vessels in both the normal and tumor microenvironment. Sprouting, the dominant means of vessel formation, is the branching of a new vessel from an established blood vessel. The other major mechanism is nonsprouting, which occurs when an existing blood vessel enlarges and splits into two separate vessels. Aggressive tumor cells may also develop microvascular channels to support neovascularization in a process known as vasculogenic mimicry. Finally, existing vasculature in the host tissue may be coopted by the tumors to increase vascular supply.
In normal tissues, the vasculature is organized and uniform in size and shape. Angiogenesis in the tumor microenvironment results in vessels that are more irregular, appearing tortuous, dilated, and leaky. The regulation of angiogenic mechanisms is provided by a complex set of growth factors that stimulate and inhibit vascular growth in response to internal and external stimuli. In general, these factors act on the cells lining the blood vessel (the endothelial cells) to regulate activity within the cellular microenvironment. In the normal cellular microenvironment, the endothelial cells are stable, dividing rarely. Pathologic angiogenesis secondary to an increase in proangiogenic factors results in endothelial cells that demonstrate unregulated division and growth. In fact, high expression of proangiogenic molecules and increased microvessel density (a marker of increased tumor vascularization) are poor prognostic factors in many solid malignancies.
Vascular Endothelial Growth Factors and Vascular Endothelial Growth Factor Receptors
Fig. 18.3 demonstrates a schematic of the VEGF pathway, a key contributor to the regulation of angiogenesis. Activation of this pathway promotes the proliferation, survival, and migration of endothelial cells leading to vascular growth. In addition, VEGF stimulation increases cell fenestration and vascular permeability, which has been associated with the development of malignant effusions in the lungs and peritoneal cavity. VEGF overexpression has been found in a majority of solid tumors, including all three major gynecologic cancers, and is associated with poor prognosis and tumor progression.

As demonstrated in Fig. 18.3 , the VEGF pathway includes seven different ligands: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, placental growth factor (PlGF)-1, and PlGF-2. The mediation of the angiogenic effects for each VEGF ligand is accomplished through one of three receptors (VEGFRs), including VEGFR-1, VEGFR-2, and VEGFR-3. These receptors belong to the class III family of tyrosine kinase receptors and are typically expressed on the vascular and lymphatic endothelium. However, these receptors may also be expressed by tumor tissue. The overexpression of VEGF in tumor cells is associated with increased tumor growth and metastasis in several solid malignancies, including ovarian and endometrial cancer. Recent studies revealed that ovarian cancers expressing VEGF receptors have higher mortality rates compared with tumors lacking VEGF receptor expression. In cervical cancer, human papilloma virus infection can lead to degradation of p53 and subsequent upregulation of hypoxia inducible factor (HIF-1). This factor can lead to increased neovascularization, supporting the rationale for antiangiogenic therapy in cervical cancer.
Activation of a given VEGFR results in subsequent downstream activation of a variety of known survival, proliferation, and migration pathways in the cell. These downstream pathways include phosphoinositide-3-kinase/akt (PI3K-AKT), Ras and Raf superfamily-MAPK (Ras-Raf-MAPK), focal adhesion kinase (FAK), and v-src sarcoma viral oncogene homolog (SRC). The activity of the VEGFR2 appears to be potentiated by the binding of co-receptors, neuropilin-1 (NRP-1) on arteries, and NRP-2 on venous and lymphatic vessels. NRP-1 and NRP-2 may have intrinsic activity through the binding of small G proteins and regulation of the cytoskeleton also.
Given the importance of the VEGF pathway, the majority of angiogenesis-related targeted therapies are focused on the VEGF family of ligands and receptors. Current options for therapy include drugs that directly target VEGF and its receptors and vascular disrupting agents that damage existing tumor related blood vessels.
Agents Targeting the Vascular Endothelial Growth Factor Pathway
Bevacizumab
Ovarian cancer.
Bevacizumab, a humanized mAb to human VEGF, was the first US Food and Drug Administration (FDA)–approved drug targeting angiogenesis. In ovarian cancer, bevacizumab has been evaluated both as a single agent and in combination therapy for primary and recurrent disease. After encouraging preclinical studies and multiple case studies, the Gynecologic Oncology Group (GOG) instituted a phase II trial of single-agent bevacizumab (15 mg/kg every 3 weeks) in persistent or recurrent refractory epithelial ovarian cancer. Despite a heavily pretreated cohort, the authors reported 13 of 62 patients (21%) experienced a clinical response, including 11 patients with partial response and two patients with a complete response. The median number of cycles was seven, and 25 patients (40.3%) had a progression-free survival (PFS) time of at least 6 months. Overall, toxicity was low, and there were no reports of bowel perforation. Another phase II trial of bevacizumab (15 mg/kg every 3 weeks) in 44 patients with ovarian cancer receiving third- or fourth-line chemotherapy demonstrated a partial response rate of 16%. The median PFS time was 4.4 months, and the median overall survival (OS) period was 10.7 months at study closure. This study was terminated early secondary to five patients (11.4%) experiencing spontaneous bowel perforation. The risk of bowel perforation appeared to be higher in patients with a higher median number of prior treatments and in whom impending bowel obstruction was suspected. Further studies are ongoing to elucidate a clear list of risk factors for perforation in the setting of bevacizumab therapy.
In the setting of relapsed disease, bevacizumab has been evaluated in combination with both cytotoxic and biologic therapies. A phase II study of bevacizumab (10 mg/kg every 2 weeks) combined with oral cyclophosphamide (50 mg/day) demonstrated a partial response rate of 24% (17 of 70 patients) at a median follow-up period of 23.2 months. The probability of being progression free at 6 months was 56% in this study. Overall toxicity was acceptable in this study. This combination has also been evaluated retrospectively at several institutions with similar encouraging results (objective response rates, 44%–53.3%). A phase II trial combining bevacizumab (15 mg/kg every 21 days) and oral erlotinib (150 mg/day) in 13 patients with recurrent Müllerian cancer showed an objective response rate of 15% and a stable disease (SD) rate of 54%. However, this trial was stopped early secondary to lack of clear benefit of the combination over single-agent bevacizumab and a higher than expected rate of bowel perforation (15%).
Success in the platinum-resistant recurrent setting as well as in several phase II trials in the upfront setting led to five major phase III trials in ovarian cancer. In the upfront setting, GOG 218 and International Collaborative Ovarian Neoplasm (ICON) 7 both include bevacizumab in combination with standard cytotoxics followed by maintenance bevacizumab. Key characteristics of these studies are summarized in Table 18.4 . Preliminary data from GOG 218 revealed a PFS benefit of 3 months in the arm that included bevacizumab treatment up front and continued as single-agent maintenance (14.1 vs. 11.2 months; hazards ratio [HR], 0.72; P <0.001). It is interesting that there was no PFS benefit in the patients who only received adjuvant bevacizumab compared with standard therapy alone. No OS benefit was noted in the arms containing bevacizumab, although this was not a primary endpoint of the study. Although different in design, ICON7 reached similar conclusions. In the arm receiving paclitaxel, carboplatin, and bevacizumab followed by bevacizumab maintenance, the PFS time was improved by 1.7 months (24.1 vs. 22.4 months; HR, 0.87; P = 0.04). OS, albeit immature, was similar between the arms.
| GOG 218 | ICON 7 | |
|---|---|---|
| Patients ( n ) | 2000 | 1520 |
| Type | Randomized | Randomized |
| Placebo-controlled | Open Label | |
| Primary endpoints | Overall survival | Progression-free survival |
| Progression-free survival | ||
| Secondary endpoints | Toxicity | Overall survival |
| Quality of life | Response rate | |
| Translational research | Biologic progression-free survival | |
| Toxicity | ||
| Quality of life | ||
| Economics | ||
| Strata | Stage (III ≤1 cm vs. >1 cm vs. IV) | Stage (I–III ≤1 cm vs. >1 cm vs. IV) |
| PS: (0 vs. 1–2) | Chemo start (≤4 wk vs. > 4) | |
| Enrolling center | ||
| Sites | 490 | 142 |
| Opened | September 2005 | April 2006 |
| Closed | June 2009 | February 2009 |
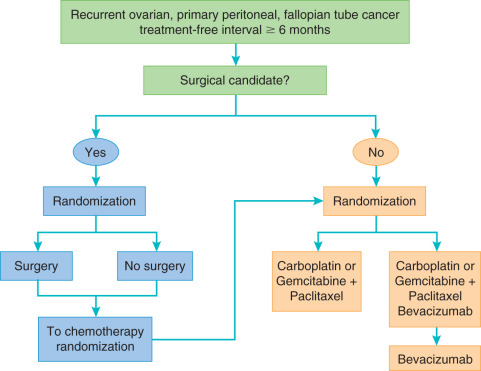
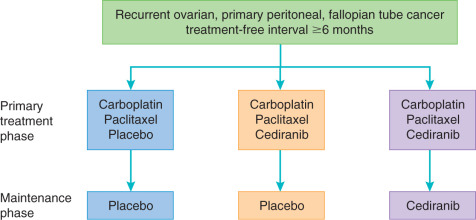
More promising results for bevacizumab have been observed in the phase III trials in the recurrent setting. GOG 213 is a bifactorial randomized study to evaluate the effect of the addition of bevacizumab to standard carboplatin and paclitaxel or gemcitabine on OS in platinum-sensitive patients ( Fig. 18.4 ). Preliminary results of this study demonstrated a significant improvement in PFS as well as a trend toward improved OS in the arm containing bevacizumab. This study is also addressing the benefit of secondary cytoreductive surgery by randomly assigning a subset of patients who meet criteria for cytoreduction to surgery versus no surgery before initiation of chemotherapy. In the OCEANS trial, patients with platinum-sensitive ovarian cancer were treated with gemcitabine and carboplatin with or without bevacizumab. This trial aimed to evaluate PFS and potential GI toxicity of this combination. Similar to GOG 213, the arm containing bevacizumab demonstrated a significant improvement in PFS of 4 months. This did not, however, yield a difference in OS. In the platinum-resistant setting, the AURELIA trial compared bevacizumab with a physician choice standard agent, including paclitaxel, liposomal doxorubicin, or topotecan, with the standard agent alone. Bevacizumab provided a statistically significant improvement in response rate and PFS when added to standard chemotherapy. Interestingly, when a subanalysis stratified by each chemotherapy cohort was performed, the greatest PFS benefit (6 months) was found in the cohort that combined weekly paclitaxel with bevacizumab. The high crossover to bevacizumab after trial participation likely influenced the lack of OS difference noted in this trial. The results of this trial yielded an FDA approval for bevacizumab in combination with chemotherapy in platinum-resistant ovarian cancer receiving one or two prior regimens.
Bevacizumab has also shown promise in nonepithelial ovarian cancer. A retrospective review of eight patients with recurrent granulosa cell tumors demonstrated a partial response rate of 38% and SD rate of 25%. This study has encouraged the development of a phase II trial by the GOG evaluating bevacizumab for women with recurrent ovarian sex cord–stromal tumors. Among 36 patients treated, 16.7% had a partial response, and 77.8% achieved SD. Certainly, these results are promising in this notoriously chemoresistant disease.
Uterine cancer.
Vascular endothelial growth factor expression has been correlated with adverse outcomes in endometrial cancer, and bevacizumab has demonstrated encouraging results in early phase clinical settings. The GOG initiated a phase II study of single-agent bevacizumab (15 mg/kg every 21 days) for advanced endometrial cancer demonstrating a 13.5% response rate and 40.4% surviving progression free at 6 months. The median OS time was 10.5 months in this trial. A subsequent trial attempted to maximize this benefit by combining bevacizumab with temsirolimus, an mTORC1 inhibitor described later in this chapter. The combination was deemed active based on reasonable response rate (24.5%) and 47% of patients surviving progression free at 6 months; however, its development has been limited by increased toxicity. In the upfront setting, the addition of bevacizumab to Intensity Modulated Radiation Therapy (IMRT) with cisplatin was evaluated in high-risk endometrial cancer. Overall, toxicity was reasonable, and the OS rate for the cohort was 96.7% at 2 years.
Cervical cancer.
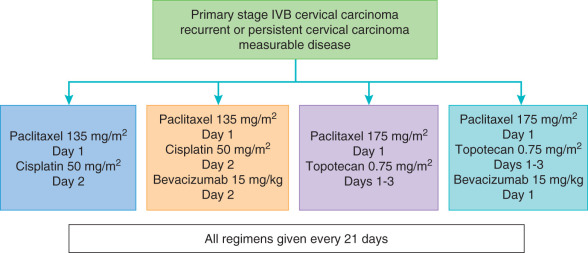
Bevacizumab is the first targeted agent that has demonstrated activity in recurrent cervical cancer. A phase II trial of single-agent bevacizumab (15 mg/kg every 21 days) had promising results in a cohort of patients with fewer than three prior regimens, achieving a median OS period of 7.29 months and acceptable toxicity. Five patients (10.9%) had a partial response, and an additional 11 patients were progression free for a minimum of 6 months. Furthermore, an analysis of six patients treated with bevacizumab in combination with 5-fluorouracil or capecitabine demonstrated a clinical benefit rate of 67%. There are several trials with bevacizumab in cervical cancer actively accruing or completed in the upfront and recurrent settings. GOG 240 combined bevacizumab with standard chemotherapy in four regimens, described in Fig. 18.5 . This study found a significantly significant improvement in OS among the arms that received bevacizumab (17.0 vs. 13.3 months, 0,71; P = 0.004), which led to an FDA approval for bevacizumab in combination with chemotherapy for advanced and recurrent cervical cancer.
VEGF-Trap (Aflibercept)
Aflibercept is a manufactured protein that acts as a decoy receptor for all VEGF-A isoforms and placental growth factor. This agent was engineered through fusion of the ligand-binding domains from two VEGF receptors with the constant region of IgG1, resulting in high-affinity VEGF binding and prevention of VEGF pathway activation. In the in vivo setting, aflibercept was found to improve ascites and reduce tumor growth.
Initial phase I trials of aflibercept for advanced solid malignancy demonstrated acceptable toxicity with clinical benefit approaching 50%. Several partial responses were observed in ovarian cancer. This led to a randomized phase II trial of aflibercept (2 mg/kg vs. 4 mg/kg) in platinum-resistant recurrent ovarian cancer. A response rate of 11% was reported with five partial responses and no mention of SD. Aflibercept has also been studied for the treatment of malignant ascites in ovarian cancer. Colombo and colleagues treated 12 patients with aflibercept (4 mg/kg) every 2 weeks and found successful prolongation in time to repeat paracentesis with minimal adverse advents. A phase I/II, multi-institutional trial reported the activity and toxicity of aflibercept in combination with docetaxel in women with recurrent ovarian cancer. Overall response in the phase II component was 54%, including 11 of 25 responders being complete. Median PFS and OS periods were 6.4 and 26.6 month, respectively. As expected, the most frequent aflibercept-associated toxicity was hypertension (11% grade 1 or 2). In endometrial cancer, the GOG performed a phase II study of aflibercept in the recurrent setting. This agent achieved a 7% response rate, and 23% of patients survived progression free for 6 months.
Agents Targeting Vascular Endothelial Growth Factor Receptors
AZD2171 (Cediranib)
Cediranib is a small molecule inhibitor of VEGFR-2, platelet-derived growth factor receptor (PDGFR), and c-kit, which has shown promise in several phase II trials. In a study of 46 patients with recurrent ovarian cancer, the clinical benefit rate of single-agent cediranib was 30%. Eight patients achieved partial response, and six patients had SD, and median PFS for the group was 5.2 months. Hirte and colleagues reported a response rate of 41% in platinum-sensitive and 29% in platinum-resistant ovarian malignancy. Toxicities in both studies included diarrhea, hypertension, mucositis, fatigue, and anorexia. Cediranib was evaluated in the upfront setting in combination with standard paclitaxel and carboplatin as part of ICON6 ( Fig. 18.6 ). Toxicity of the combination was well tolerated and yielded a PFS benefit of 3 months. Importantly, the use of cediranib in the maintenance setting was not associated with a significant improvement in OS. This combination of paclitaxel and carboplatin with cediranib was studied in cervical cancer as well, yielding 2 months of increased PFS at the expense of increased toxicity. Single-agent cediranib in endometrial cancer had promising activity, with a 12% response rate and 30% of patients progression free at 6 months, leading to the development of an ongoing combination trial with standard of care chemotherapy.
IMC-1121B (Ramucirumab)
IMC-1121B is the most developed fully humanized mAb to VEGFR-2. A phase I trial in 12 patients with advanced solid malignancies demonstrated a clinical benefit in nine patients (two partial responses, seven with SD). These promising results led to a phase II trial of ramucirumab in recurrent ovarian cancer. This study revealed that single-agent ramucirumab (8 mg/kg) only achieved a response rate of 6% with 56.7% of patients having SD. Thus, there has been minimal effort to explore this agent further in gynecologic malignancies. Several other mAbs to VEGF receptors are still in development, including IMC-18F1 and IMC-1C11.
Agents Targeting Multiple Vascular Endothelial Growth Factor–Related Molecules
Sunitinib
Sunitinib is an oral receptor tyrosine kinase inhibitor whose targets include VEGFR, PDGFR, epidermal growth factor (EGF), and the stem cell factor (KIT) receptor. This drug has been evaluated in the treatment of recurrent ovarian cancer in several phase II trials and one phase III trial. A phase II trial of sunitinib (50 mg/day intermittent dosing, 4 of 6 weeks vs. 37.5 mg/day) in recurrent platinum-sensitive and platinum-resistant ovarian cancer demonstrated a 66% clinical benefit with partial response in one patient, CA125 responses in three patients, and SD in 16 patients. Of note, responses were only seen in patients in the intermittent cohort. Common side effects were hand and foot reaction, fatigue, hypertension, and mucositis. Mackay and colleagues reported the results from a phase II trial of sunitinib (50 mg/day) in 19 patients with advanced or metastatic cervical cancer. Although they achieved no objective responses, 16 patients achieved SD with a median duration of 4.4 months. There are several ongoing active phase II trials of single-agent sunitinib in endometrial and cervical cancer and clear cell ovarian cancer (GOG 254).
Pazopanib
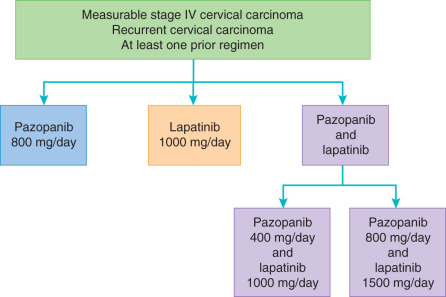
Pazopanib inhibits all of the VEGF receptors (VEGFR1, VEGFR2, and VEGFR3) and PDGFR-α and -β and the KIT receptor. This small molecule inhibitor (800 mg/day) has been evaluated in a phase II study of 36 patients with recurrent ovarian cancer by CA-125 and nonbulky disease. This study revealed a 31% response by CA-125 level and a 56% SD rate. Among the 17 patients with measurable disease, 18% had a partial response. Ongoing ovarian cancer studies of pazopanib include combination with liposomal doxorubicin in the recurrent setting and in combination with paclitaxel and carboplatin in the upfront setting. A phase III, placebo-controlled study of pazopanib for consolidation after completion of primary chemotherapy in ovarian cancer was recently published. Use of pazopanib as maintenance therapy prolonged PFS by 5.6 months when compared to placebo. Adverse events were increased in the pazopanib arm, with 33% of patients terminating this agent early. Interim results did not yield a subsequent benefit in OS. Interestingly, a subset analysis of East Asian women included in this study revealed a negative impact of pazopanib on PFS among this cohort. Further studies are ongoing to understand the mechanism underlying this difference. Pazopanib has also been combined with chemotherapy in the recurrent ovarian cancer setting. One randomized phase II trial comparing weekly paclitaxel with or without pazopanib revealed a 3-month PFS benefit in the setting of moderately increased adverse events, including neutropenia and fatigue. However, another placebo-controlled trial failed to demonstrate a benefit with the addition of pazopanib to weekly paclitaxel in recurrent ovarian cancer. In cervical cancer, pazopanib was explored alone and in combination with lapatinib (a small molecule EGFR inhibitor to be discussed later) in the treatment of advanced and recurrent disease ( Fig. 18.7 ). The combination pazopanib and lapatinib arm was closed early after a futility analysis, leaving the randomized phase II trial to compare pazopanib with single-agent lapatinib. Both progression-free (HR, 0.66; 90% CI 0.48–0.91) and OS (HR, 0.67; 90% CI 0.49–0.99) were superior in the monotherapy pazopanib arm. Median OS was 50.7 weeks versus 39.1 weeks for pazopanib and lapatinib, respectively.
Brivanib
Brivanib alaninate is a prodrug of brivanib, a dual inhibitor of VEGFR2 and FGFR. This compound has demonstrated acceptable toxicity and activity when given alone or in combination, in multiple solid tumors, including hepatocellular carcinoma refractory to other antiangiogenic therapies. Brivanib was evaluated in a phase II study for recurrent or persistent endometrial cancer and found worthy to pursue further based on a response rate of 19% (one complete response and seven partial responses) and 30% of patients surviving progression free at 6 months. Similar favorable activity was found in the first stage of a phase II trial of brivanib in recurrent cervical cancer; however, the trial was closed early because of drug availability.
Nintedanib
Nintedanib is a multikinase inhibitor targeting three key angiogenic receptors: VEGFR, PDGFR, and FGFR. A phase I trial of this agent in patients with gynecologic malignancies revealed a promising response rate, with five of seven patients with measurable disease demonstrating response and two achieving SD. Nintedanib (250 mg/day) was evaluated as a maintenance therapy compared with placebo in recurrent ovarian cancer after response to standard therapy. Although the trial was not powered to compare the two arms, PFS was less in the placebo arm (2.8 months) compared with the nintedanib arm (4.8 months). A randomized phase III placebo-controlled trial compared standard paclitaxel and carboplatin with or without nintedanib in previously untreated primary ovarian cancer patients. The combination arm was associated with significantly prolonged PFS, although clinically, this was a median increase of approximately 2 weeks. Furthermore, there was significant toxicity in the combination arm, including GI and hematologic adverse events. A phase II study of this agent in recurrent endometrial cancer did not provide sufficient activity to warrant further study as a single agent.
Vascular Disrupting Agents
This broad group of antiangiogenic drugs acts to occlude preexisting vasculature in the tumor rather than prevent neovascularization. The disturbance of existing vessels leads to ischemia; hemorrhagic necrosis; and ultimately, cellular death. Of note, these agents are able to selectively target tumor blood vessels by taking advantage of the differences between normal and tumor endothelial cells. Two major types of vascular disrupting agents exist: small molecule based and ligand based. The majority of vascular disrupting agents under evaluation in gynecologic cancers target small molecules.
Vadimezan (ASA404/DMXAA)
Vadimezan, 5,6-dimethylxanthenone-4-acetic acid, is a flavone acetic acid analog that increases production and release of tumor necrosis factor α. This leads to endothelial cell apoptosis and decreased perfusion of the tumor. Gabra and colleagues evaluated this drug in combination with paclitaxel and carboplatin for the treatment of recurrent ovarian cancer. The arm that received vadimezan conferred significant improvement in response rate compared with the control arm (64% vs. 49%) without additional adverse effects.
Fosbretabulin (Combretastatin A4); Ombrabulin (AVE8062)
Fosbretablin is a tubulin-binding agent that causes vascular congestion and decreased tumor blood flow by changing endothelial cell shape. This effect is very rapid, occurring within 1 hour of drug administration. This agent has been evaluated primarily for the treatment of platinum-resistant ovarian cancer. Treatment with combretastatin A4 (63 mg/m 2 ) in addition to paclitaxel (175 mg/m 2 ) and carboplatin (area under the curve [AUC] 5) resulted in a 13.5% response rate by Response Evaluation Criteria for Solid Tumors (RECIST) and a 34% response rate by Gynecologic Cancer Inter Group (GCIG) criteria without additional observed toxicity. A randomized phase II trial of bevacizumab with or without fosbretabulin demonstrated higher response rate as well as improved PFS in the combination arm among patients with platinum-resistant ovarian cancer. OS data from this study have not yet been presented. Ombrabulin is a combrestatin derivative that has shown excellent efficacy in preclinical ovarian cancer models. This agent appears to have a similar toxicity profile to combretastatin A4 and is undergoing evaluation in a phase II trial in combination with paclitaxel and carboplatin in platinum-sensitive recurrent ovarian cancer (OPSALIN).
Other Antiangiogenic Agents
Thalidomide
Thalidomide is a legacy agent that has found recent renewed interest as an antiangiogenic agent. Thalidomide inhibits FGF-2 by interacting with the FGF-2 receptor. In addition, this agent acts as an immune modulator by inducing production of interferon-γ, interleukin-2 (IL-2), and IL-10. Thalidomide (200 mg/day) added 4 months to OS when added to topotecan (1.25 mg/m 2 ) for the treatment of recurrent ovarian cancer. A phase II of thalidomide (200–1000 mg/day) in persistent or recurrent endometrial cancer demonstrated a 25% clinical benefit with two partial responses and four patients with disease stabilization. The GOG reported randomized phase III trial results comparing thalidomide (200 mg/day) with tamoxifen (20 mg/day) for biochemically recurrent ovarian carcinoma. In this study, there was essentially no difference in PFS, with thalidomide achieving 3.2 months compared with 4.5 months for tamoxifen. Patients on the thalidomide arm had an increased risk of death (HR, 1.76; 95% CI 1.16–2.68). Furthermore, patients receiving thalidomide had a higher incidence of grade III and IV toxicity. Common toxicities of thalidomide include hematologic, GI, and cardiovascular complications. Lenalidomide (Revlimid) is a thalidomide derivative that demonstrated modest activity in platinum resistant ovarian cancer, yielding a clinical benefit rate of 38% at 4 months, including four partial responses by RECIST.
Phosphatidylinositol-3-Kinase/AKT Pathway
The PI3K/AKT pathway plays a central role in cell survival, growth, and avoidance of apoptosis. Fig. 18.8 demonstrates a simple schematic of this complex pathway that is known to interact with many other cellular growth and survival pathways. This pathway may be activated by a large number of receptor tyrosine kinases, including the EGFR family and the insulin-like growth factor receptors (IGFRs). Thus a variety of mitogenic substances are involved in its activation.
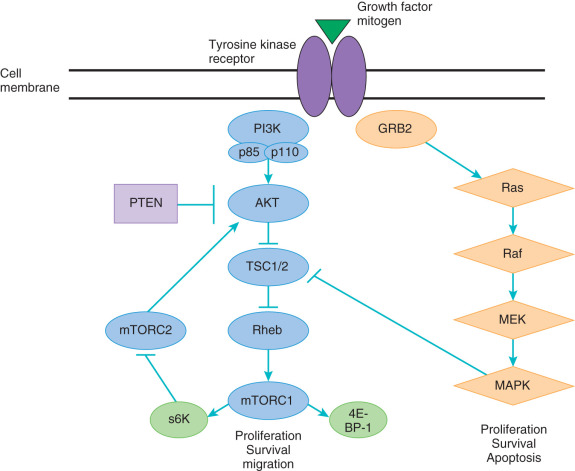
Activation of the pathway starts with the PI3K family, which consists lipid and serine/threonine kinases composed of heterodimers, including a catalytic and regulatory subunit. Activation of PI3K leads to phosphorylation of phosphatidylionositol-4,5-bisphospate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 acts as a second messenger to bind a variety of targets and recruit them to the plasma membrane, leading to their activation. One critical downstream mediator of PIP3 is AKT, which on activation acts on a number of different targets that directly affect cellular survival, proliferation through activation of transcription and translation, evasion of apoptosis, and resistance to chemotherapy. One key downstream target of AKT is the mammalian target of rapamycin (mTOR), a serine/threonine kinase. The upregulation of mTOR by AKT leads to activation of downstream regulator protein S6 kinase, which directly affects protein translation and the progression of growth through the cell cycle.
The PI3K/AKT pathway is known to be activated in a variety of cancers, especially in gynecologic malignancies. The phosphatase and tensin homolog on chromosome 10 (PTEN) is a tumor suppressor that encodes for a serine/threonine kinase, which acts directly to dephosphorylate PIP3 to PIP2. In patients with PTEN mutation and loss of function, there is an overaccumulation of PIP3, leading to constitutive activation of the AKT pathway. The PI3K/AKT pathway is also frequently activated through mutations in PIK3CA, which encodes for the activating subunit (110α) of PI3K or through mutations in AKT.
As described in Chapter 19 , the aforementioned mutations are commonly found in endometrial cancer and less frequently in the other gynecologic malignancies. Furthermore, this pathway is thought to be targeted more frequently in cancer than any other pathway aside from p53. Thus, PI3K/AKT signaling provides a promising target for the treatment of malignancy and is under active exploration. Currently, the drugs targeting this pathway consist primarily of small molecule inhibitors of key pathway components. The inhibition of only one member of the pathway may not be sufficient to affect tumor growth given the significant pathway cross-talk and feedback loops. For example, mTOR is known to regulate cellular growth and proliferation through activation of several downstream proteins. These proteins also participate in a feedback loop that can lead to subsequent upregulation of AKT phosphorylation. Thus, the exploration of combination therapies in this pathway is paramount.
Agents Targeting mTOR
mTOR is a key downstream protein of the PI3K/AKT pathway, consisting of two major complexes (mTORC1 and mTORC2). Initial attempts at targeting this pathway node were directed at mTORC1 (rapalogs). Overall, trials in gynecologic cancer with rapalogs have demonstrated only modest success. Thus, there are newer agents in development that compete with ATP on the mTOR catalytic site and inhibit mTORC1 and mTORC2.
Temsirolimus (CC1-779)
Temsirolimus (25 mg intravenously weekly), a water-soluble ester of rapamycin, had a favorable response rate in a phase II trial of patients with recurrent or metastatic endometrial cancer treated with a maximum of one prior regimen. Among the chemotherapy naïve cohort, 14% of 29 evaluable patients had a partial response, and an additional 69% achieved SD. However, among patients treated with prior chemotherapy, only 4% had partial responses, and 48% had SD of median 4 months’ duration. A study of temsirolimus in 54 heavily pretreated patients with recurrent endometrial cancer yielded 9% partial responses and 24% progression free at 6 months. Temkin and colleagues reported on a phase I trial of temsirolimus combined with topotecan to treat a variety of advanced and recurrent gynecologic malignancies. The drug combination was well tolerated in patients without a history of radiotherapy and achieved SD in 9 of 11 patients at 8 weeks. Trials incorporating temsirolimus have shown manageable toxicities, including hypertriglyceridemia, hyperglycemia, electrolyte abnormalities, and rash.
This agent has been combined with a variety of combination chemotherapies and targeted therapies in endometrial cancer. As noted earlier, the combination of temsirolimus with bevacizumab did not yield a significant increase in response or PFS at 6 months to warrant further exploration. Results of this combination in ovarian cancer were similar as well. To improve on the current standard of care for advanced and recurrent chemotherapy-naïve endometrial cancer, the GOG performed a randomized phase II trial with three arms: (1) temsirolimus in combination with paclitaxel and carboplatin; (2) bevacizumab, paclitaxel, carboplatin; and (3) ixabepilone, paclitaxel, and carboplatin. This trial did not demonstrate a significant increase in PFS in any of the arms compared to historical controls. Interestingly, the secondary endpoint of OS was increased in the bevacizumab arm (HR, 0.71; 92% CI 0.55–0.91 compared with historical controls). In recurrent cervical cancer, single-agent temsirolimus had similar modest effects, achieving only one (3%) partial response and a 6-month PFS rate of 28%. A phase II trial combining temsirolimus with carboplatin and paclitaxel was recently completed in advanced clear cell carcinoma of the ovary; results are anticipated soon.
Everolimus (RAD001)
Everolimus is an orally bioavailable ester that is a potent inhibitor of mTOR. Two phase II studies of everolimus (10 mg/day) as a single agent in recurrent endometrial cancer have been reported, with only modest activity. Slomovitz and colleagues demonstrate a short- and long-term clinical benefit rate with 43% and 21% of patients achieving SD at 8 weeks and 20 weeks, respectively. The ENDORAD study of 54 metastatic endometrial cancer patients reported five partial responders and a median PFS period of only 2.8 months. Given these results, attempts have been made to maximize the success of this drug in combination with other targeted agents, traditional cytotoxic chemotherapy, and hormones. The latter combination has gained keen interest in breast and endometrial cancers because endocrine resistance may be mediated by activation of the PI3K pathway. A phase II study of the combination of everolimus and letrozole markedly improved on the single-agent efficacy, with confirmed objective response rate of 32%, including nine complete responders. Furthermore, the clinical benefit rate was 40%, and the combination was well tolerated.
Ridaforolimus (AP23573; MK-8669)
Ridaforolimus, previously known as deforolimus, is an mTOR inhibitor that may be given orally or intravenously. In a phase II study of ridaforolimus (12.5 mg intravenously daily for 5 days every other week) for patients with advanced or recurrent endometrial cancer, 13 of 45 patients achieved clinical benefit, including five with partial response. The oral formulation (40 mg orally daily for 5 days every 3 weeks) was evaluated in recurrent endometrial cancer as well. Similar to the intravenous (IV) formulation, ridaforolimus yielded a response rate of 8.8% and disease stabilization of 53%. Final results of a multi-institutional randomized phase II trial of ridaforolimus (40 mg orally for 5 days every 28 days) in women with recurrent, previously treated endometrial cancer were reported in 2015. The control arm of the trial was the physician’s choice of either hormones or chemotherapy. Overall, 130 patients (114 evaluable) were enrolled—64 to ridaforolimus, 52 to hormonal therapy, and 13 to chemotherapy. The response rates in each arm were similar (0% vs. 4% for ridaforolimus and control participant, respectively). However, PFS was significantly longer in the ridaforolimus arm (1.7 months) when assessed by either the investigators or by independent radiologic review. OS has not been reported.
Agents Targeting AKT
Given the modest activity of targeting downstream regulators of the PI3K/AKT pathway, there is interest in targeting higher level nodes of the pathway. There are several AKT-inhibiting agents in development and the majority are undergoing evaluation in phase I trials. MK-2206 is a highly selective non-ATP competitive allosteric Akt inhibitor that is equally potent against Akt1 and Akt2 and demonstrated efficacy in vitro and in vivo in several tumor models. A phase I dose escalation study in patients with solid tumors has been completed to identify tolerance of the compound at 60 mg orally every other day. Dose-limiting toxicities were skin rash, mucosal inflammation, and hyperglycemia. Three ovarian cancer patients treated in this study had reducing CA-125 values. A single-agent study of MK-2206 in recurrent endometrial cancer stratified by presence of PI3KCA mutation had only modest response rates (6%) and little overall clinical benefit.
Agents Targeting PI3K
Although several companies are exploring the inhibition of PI3Kinase in solid tumors, there are only two PI3K inhibitors in phase II trials. Pilaralisib is a highly selective oral inhibitor of PI3K and successfully inhibits tumor growth in vivo. A phase I trial revealed acceptable toxicity and durable clinical benefit. A phase I trial combining pilaralisib with carboplatin and paclitaxel incorporated a dose expansion for ovarian and endometrial cancer patients secondary to favorable responses in those tumor types, although final results have not yet been reported. In recurrent endometrial cancer, pilaralisib as a single agent yielded only a 6% response, and the rate of PFS at 6 months was only 12%.
Enzastaurin is an oral multikinase inhibitor that primarily acts to suppress tumor growth through the inhibition of PI3K. A phase I study of enzastaurin combined with bevacizumab revealed promising responses in advanced solid tumors, especially among ovarian cancer (29% partial response or complete response). Furthermore, 51% of 21 ovarian cancer patients remained in the study for greater than 6 months. A phase II study of enzastaurin alone in recurrent ovarian cancer had limited activity, with a response rate of only 7.4% (D. Armstrong, personal communication). These results led to the development of several phase II trials of enzastaurin in combination with standard chemotherapy for advanced ovarian cancer. Vergote and colleagues reported that the combination was well tolerated, however, did not yield a statistically significant improvement in progression free survival.
Combination Agents
In addition to the agents targeting specific aspects of the PI3K/AKT pathway noted previously, there are a large number of combination drugs in development and in early clinical trials. These include combination PI3K/mTOR, PI3K/AKT, and PI3K/MEK inhibitors. Given the extensive cross-talk and feedback loops involved in this pathway, the inhibition of two major nodes in one or more pathways is rational and may lead to improved outcomes. Unfortunately, early reports from single-agent studies of PI3K/mTOR inhibitors have not yielded increased efficacy over the single node agents reported earlier. Furthermore, the majority of published studies combining PI3K/AKT pathway inhibitors with MEK inhibitors have reported limited activity secondary to significant toxicities found with the combinations. A large study of the combination of SAR245409 (PI3K/mTOR inhibitor) with pimasertib (MEK inhibitor) is ongoing in low-grade serous ovarian cancer.
Ras/Raf Pathway
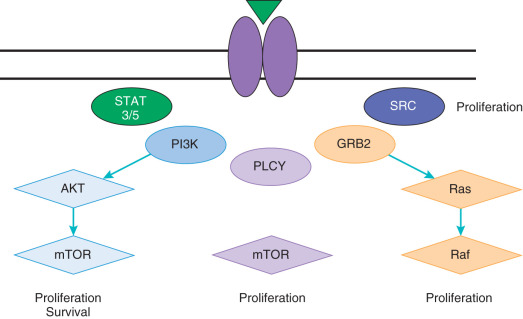
The Ras/Raf pathway is an essential regulator of tumorigenesis, including regulation of cell growth, survival, death, and motility ( Fig. 18.9 ). This pathway is tightly linked to the PI3K/AKT pathway, creating series of feedback loops that can lead to chemotherapy and targeted therapy resistance. Furthermore, this pathway is often activated in tandem with the PI3K/AKT pathway because it shares activating mitogens and receptors, including the EGFR family. Ras is a GTPase that is activated by cell surface signaling and subsequently phosphorylates downstream extracellular signal-related kinases (ERKs). Two essential pathway kinases are Raf and MEK, which ultimately serve to activate numerous downstream regulators and drive tumorigenic processes. Activation of the Ras/Raf pathway can occur through activating mutations in KRAS, HRAS, BRAF , and MEK . Ras mutations are the most common across solid tumors; however, targeting these proteins has been difficult. Thus, agents directed at downstream targets such as MEK and ERK are the most well developed.
Agents Targeting MEK
The highest rates of Ras/Raf pathway aberrations are found in low-grade serous and mucinous ovarian cancer as well as endometrial cancer. The rarity of mucinous cancer has limited the exploration of targeted agents for this indication. However, MEK inhibitors have been quite successful for the treatment of low-grade ovarian cancer. Selumetinib was explored in a phase II trial by the GOG, yielding a 15% objective response rate and a 65% SD rate in this notoriously chemo-insensitive disease. Based on the results of this trial, two large phase III studies are ongoing to compare MEK inhibition therapy with physician choice chemotherapy in recurrent low-grade serous ovarian cancer. In recurrent endometrial cancer, single-agent results for MEK inhibition have been limited. A phase II GOG trial of selumetinib reported a 6% objective response ( n = 3) and an event-free survival rate at 6 months of 12%. As noted earlier, these modest results have led to exploration of MEK inhibitor combinations for endometrial cancer.
Poly-ADP-Ribose Polymerase Pathway
DNA damage can be repaired through a variety of pathways in the cell, including base excision, direct repair, mismatch repair, and nucleotide excision repair. High activity of DNA damage repair pathways is a known mechanism of resistance to cytotoxic chemotherapy. Poly-ADP-ribose polymerase (PARP) is a nuclear enzyme that contributes to the repair of single-stranded breaks in DNA along the base excision repair pathway ( Fig. 18.10 ). There are several key mechanisms of action of this therapy. When PARP is inhibited, accumulation of single-stranded breaks can lead to double-stranded breaks and ultimately, cellular death. The use of PARP inhibitors in carriers of BRCA mutations is effective because tumor cells in these patients have dysfunctional homologous recombination repair, a primary double-stranded break repair mechanism. Furthermore, PARP inhibitors also stimulate a separate DNA repair mechanism, the nonhomologous end joining (NHEJ) pathway, which is error prone and yields cell death. Finally, PARP inhibitors appear to gain antitumor function through PARP “trapping” in which the PARP protein is trapped on the DNA complex, interfering with further DNA repair and replication. Of note, some BRCA mutation carriers appear resistant to PARP inhibition. Proposed mechanisms of PARP inhibitor resistance include the presence of secondary BRCA2 mutations that restore BRCA function, activation of Rad51, loss of 53BP1 leading to suppression of the NHEJ pathway, and increased activity of drug efflux pumps. A better understanding of these mechanisms will no doubt lead to expansion of the use of PARP inhibitors alone and in combination for the treatment of gynecologic malignancies.

PARP inhibitors have been primarily explored in ovarian cancer given the strong correlation between BRCA mutations and this disease. The potential indication of PARP inhibitor use is actively expanding to tumors with homologous recombination deficiency, including those with BRCA1 promotor methylation, aberrations in ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR) and the Fanconi anemia pathway. Certainly, preclinical data supporting the consideration of PARP inhibition in other gynecologic malignancies exist, including the similar genomic landscape found in a subset of endometrial cancers based on molecular data generated from The Cancer Genome Atlas, indicating genomic instability and homologous recombination deficiency. In addition, the presence of PTEN alterations in endometrial cancer may increase sensitivity to PARP inhibition, based on work in cell lines as well as a case report. Similarly, preclinical data have supported potential synergy between PARPs in combination with chemotherapy or with radiation in cervical cancer cell lines. Thus, early-stage trials to evaluate PARP inhibition in both of these tumor types have been initiated.
Olaparib
Olaparib is the most studied PARP inhibitor to date in ovarian cancer. Preclinically, this orally active PARP inhibitor induces synthetic lethality in cells deficient in BRCA . In a study of two doses of olaparib in BRCA -mutant patients with advanced ovarian cancer, both doses were well tolerated and demonstrated modest effect. Olaparib at 100 mg twice a day (bid) achieved a response rate of 12.5% and a clinical benefit rate of 16.7% among 24 patients. The 400-mg bid dose had higher levels of response and clinical benefit among 33 patients, 33% and 57.6%, respectively. Additional phase II studies of olaparib in recurrent high serous ovarian cancer have confirmed favorable response in patients with and without BRCA mutations. Olaparib also appears to reintroduce chemosensitivity to carboplatin and paclitaxel in heavily pretreated patients.
Although olaparib has been well tolerated as a single agent with primarily adverse events consisting of fatigue, nausea, and vomiting, early studies have indicated that there may be increased toxicity in combination with cytotoxic chemotherapy, necessitating dose reduction. Oza and colleagues compared the combination of olaparib with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in platinum-sensitive recurrent ovarian cancer. This study included a maintenance arm of olaparib alone. Although there was no significant difference in response rate, there was improvement in PFS in the olaparib arm, which became clear during the maintenance phase of the trial. An additional study confirmed that olaparib monotherapy as maintenance after platinum-based chemotherapy for platinum-sensitive recurrence significantly increased PFS, especially in women with BRCA mutations.
Whether this class of compounds outperforms chemotherapy in selected germline BRCA mutation carries is unclear. A randomized phase II trial compared olaparib (200 mg bid and 400 mg bid) with pegylated liposomal doxorubicin (50 mg/m 2 ) in BRCA patients progressing within 12 months of platinum. The primary endpoint was PFS. Although response rates were numerically higher for the olaparib arms, PFS was no different between the arms. This may be due to higher than expected efficacy of liposomal doxorubicin in BRCA- mutant ovarian cancer patients.
Activity as a single agent in a platinum-resistant setting led to the FDA approval of olaparib in a tight niche, including those treated for ovarian cancer with more than three prior chemotherapies for recurrent disease. The approval of olaparib for BRCA -related ovarian cancer marks the first therapy approved for gynecologic malignancies with an associated biomarker. Olaparib has also achieved registration in Europe as a maintenance therapy after response to platinum-therapy after platinum-sensitive recurrence. Additional phase III trials to evaluate the role of olaparib as maintenance therapy in BRCA -mutant ovarian cancer in the upfront or platinum-sensitive recurrent setting have either completed accrual or are ongoing.
Veliparib
Veliparib, an oral inhibitor of PARP-1 and PARP-2, has also demonstrated encouraging activity in recurrent ovarian cancer. A phase 2 study of veliparib as a single agent yielded a response rate of 26% in women with BRCA- mutant ovarian cancer. Adverse events were modest and expected, consisting primarily of hematologic toxicities. Thus far, compared with other PARP inhibitors, this agent has been the most successfully combined with cytotoxic agents. However, it is unclear what benefit the addition of veliparib may add to cytotoxic chemotherapy. A randomized phase II trial of oral cyclophosphamide with veliparib did not find additional benefit in terms of response rate when compared with cyclophosphamide alone. A subsequent large scale phase 1 trial identified a tolerable recommended phase II dose of veliparib in combination with paclitaxel, carboplatin, and bevacizumab therapy in upfront advanced ovarian cancer. The combination including maintenance veliparib is now under study as part of a phase III clinical trial of the NRG Oncology group (GOG 3005).
Rucaparib
Preclinical studies of rucaparib have revealed that this agent is a potent inhibitor of PARP-1 and -2 with significant activity in ovarian cancer. The phase 1 trial of the single agent discovered a phase II dose that is well tolerated, with toxicities primarily including neutropenia, anemia, and elevated liver enzymes. A subset analysis of four patients with ovarian and primary peritoneal cancer reported 80% with objective response leading to the ongoing ARIEL2 study in women with platinum-sensitive recurrent ovarian cancer. An objective of this trial is to evaluate a genomic signature to predict response to PARP inhibition in pretreatment tumor tissue. BRCA -mutant tumors had impressive overall response rate of 69% and median PFS of 9.4 months. Conversely, BRCA negative tumors had the lowest evidence of activity with response rate and median PFS of 13% and 3.7 months, respectively. BRCA-like tumors included those with evidence of genome-wide loss of heterozygosity in the absence of BRCA mutation. Interestingly, these tumors had evidence of efficacy with response rates of 30% and median PFS of 7.1 months. Rucaparib is currently under evaluation as a switch maintenance therapy in the platinum-sensitive recurrent setting after response to chemotherapy in the ARIEL3 trial.
Niraparib
Similar to the aforementioned agents, niraparib inhibits PARP-1 and PARP-2 across a variety of advanced solid tumors. The phase 1 trial identified a recommended phase II dose of 300 mg/day, with the most common adverse events including myelosuppression, fatigue, and GI toxicity. The trials included BRCA- mutant and proficient tumors, achieving objective response rates of 40% and 16%, respectively. After these promising results, niraparib is undergoing evaluation in similar setting to rucaparib (ARIEL3) and olaparib (SOLO-2). A single-agent study of niraparib (QUADRA) in women who have received at least three prior lines of chemotherapy is ongoing for BRCA -mutant ovarian cancer. This agent is also under evaluation in the maintenance setting as well as in combination with immune therapies.
Epidermal Growth Factor Receptor Pathway
The EGFR pathway was identified as an anticancer therapy target secondary to myriad genetic alterations found in a variety of solid tumor types, including ovarian, endometrial, and cervical carcinomas. The pathway consists of four tyrosine kinase cell-surface receptors, including EGFR (ErbB-1), HER2/neu (ErbB-2), Her-3 (ErbB-3), and Her-4 (ErbB-4). Each receptor is specific for a variety of ligands, including EGF, transforming growth factor α (TGF-α), and neuregulins (NRGs) ( Table 18.5 ). Although the receptors are activated through a traditional tyrosine kinase receptor mechanism, they also require the union between two identical receptors (homodimerization) or two different receptors within the same family (heterodimerization) for downstream activation.
| EGF Receptor | Ligands |
|---|---|
| EGFR (ErbB-1) | EGF |
| Transforming growth factor α | |
| Amphiregulin | |
| Epigen | |
| Betacellulin | |
| Heparin-binding growth factor | |
| Epiregulin | |
| HER2/neu (ErbB-2) | None |
| HER-3 (ErbB-3) | Neuregulin 1 |
| Neuregulin 2 | |
| HER-4 (ErbB-4) | Betacellulin |
| Heparin-binding growth factor | |
| Epiregulin | |
| Neuregulin 1 | |
| Neuregulin 2 | |
| Neuregulin 3 | |
| Neuregulin 4 | |
| Tomoregulin |
Upon activation, the EGFRs induce activation of a variety of cell survival pathways, including the Ras/Raf/MEK and PI3K pathways (see Fig. 18.9 ). In addition, EGFRs are known to negatively affect apoptosis and induce invasion. EGFRs are also present on endothelial cells within the tumor microenvironment. EGF ligands demonstrate a direct effect on the endothelial cells, leading to increase in endothelial cell proliferation and angiogenesis. Thus, targeting this pathway has been an area of significant research for the treatment of gynecologic malignancies. Unfortunately, recent studies of drugs affecting the EGFR pathway have only demonstrated modest success in gynecologic cancer. This may be explained, in part, by research that demonstrates that EGFR has a role in glucose transport in cancer cells. EGFR associates with the sodium/glucose cotransporter (SGLT1) to promote the uptake of glucose into cancer cells. Of note, the action appears to be kinase independent. Thus, the blockage of kinase activity with antibodies or small molecules may not be sufficient to make a significant impact on tumorigenesis. Furthermore, based on data in multiple tumor types, response to EGFR inhibitors appears to be correlated with mutation status rather than expression. The rate of EGFR mutations in gynecologic malignancies is quite low; thus, the potential for success of these compounds as a single agent is unclear.
Small Molecule Inhibitors Targeting Epidermal Growth Factor Receptor
Gefitinib
Gefitinib is a small molecule inhibitor that prevents EGFR activation through binding the ATP-binding site of the receptor. This agent has been evaluated extensively in ovarian cancer with unsatisfactory results. Gefitinib (500 mg/day) was evaluated in a phase II trial in 30 patients with recurrent or persistent ovarian cancer treated with up to two prior regimens. Of 27 evaluable patients, four had SD for more than 6 months, and only one patient had an objective response. Of note, patients with EGFR expression in their tumors had longer median PFS periods, and the one objective response was found in a patient with a mutation in EGFR. This suggests that the use of mutation status to guide targeted therapies may provide better response rates.
Gefitinib (500 mg/day) has also been evaluated in combination with carboplatin (AUC 5) and paclitaxel (175 mg/m 2 ) for platinum-sensitive and platinum-resistant recurrent ovarian cancer. Nineteen percent of platinum-resistant and 62% of platinum-sensitive patients had responses to this combination therapy. OS was also acceptable, reaching 17 months for the resistant group and 26 months for the sensitive group. However, there appeared to be more hematologic toxicity compared with the standard regimen. The combination of gefitinib (500 mg/day) with tamoxifen (40 mg/day) in recurrent ovarian cancer required dose reduction secondary to diarrhea in 10 of 56 patients and resulted in no objective responses. However, 16 patients achieved SD, and the median survival period was 253 days.
The only published trial of gefitinib (500 mg/day) in cervical cancer reported minimal activity. Of 28 evaluable patients, there were no objective responses, and 20% achieved SD for a median duration of 112 days. It is interesting that disease control was not associated with the expression of EGFR in tumor specimens. Similarly, a study of gefitinib as a single agent in recurrent endometrial cancer achieved only one responder (4%), and four patients were progression free at 6 months (15%). EGFR mutation, EGFR expression, and hormonal expression were not associated with survival in this study.
Erlotinib
Erlotinib (150 mg/day) is reversible inhibitor of EGFR, acting to block autophosphorylation of the tyrosine kinase portion of the receptor. Based on overexpression of EGFR and preclinical results that indicated that the combination of EGFR-directed therapies and chemotherapy would potentially be synergistic, a phase II trial of erlotinib in combination with standard paclitaxel (175 mg/m 2 ) and carboplatin (AUC 6) for first-line treatment in ovarian cancer was performed. The primary endpoint was pathologic complete response rate with a plan to compare with historical control participants. Of a total of 56 patients treated, erlotinib in combination with standard chemotherapy achieved a pathologic complete response rate in 11 patients, which was not a significant improvement over historical control participants. Erlotinib was also explored as a maintenance therapy in patients with no sign of progression after frontline treatment for ovarian cancer in a large randomized control trial. After 2 years of therapy, there was no difference in PFS or OS between the erlotinib and observation arms.
In the setting of recurrent ovarian cancer, erlotinib has been primarily evaluated in combination with other therapies, although a solo trial is ongoing. Erlotinib in combination with carboplatin (AUC 5) achieved a response rate of 57% in the platinum-sensitive arm and 7% in the platinum-resistant arm with no unexpected toxicities. As noted previously, a trial of erlotinib with bevacizumab (15 mg/kg) was terminated early secondary to a high level of bowel perforation. The use of erlotinib as a single agent in chemotherapy-naïve endometrial cancer was well tolerated, and four of 32 patients had an objective response. In addition, 15 patients had SD lasting a median of 4 months. EGFR mutations were evaluated and did not correlate with response.
Schilder and colleagues reported disappointing results of erlotinib as a single agent in cervical cancer, with only one of 25 patients achieving objective response and four with SD. Only one patient in this trial had PFS period longer than 6 months. The combination of erlotinib with cisplatin (40 mg/m 2 weekly) with radiotherapy in locally advanced cervical cancer was found to be feasible, with acceptable toxicity and a complete response rate of 91%.
Monoclonal Antibodies Targeting Epidermal Growth Factor Receptor
Cetuximab
Cetuximab (250 mg/m 2 ) is a chimerized mAb to the ligand-binding domain EGFR, which has been evaluated as a treatment for primary and recurrent ovarian cancer as a single agent and in combination with traditional cytotoxics. As a single agent in 25 patients, cetuximab achieved only one partial response and nine patients with SD. The median PFS period was only 2.1 months. Cetuximab in combination with carboplatin (AUC 6) in 28 patients with recurrent platinum-sensitive ovarian cancer also had disappointing results, with only nine objective responses and eight patients with SD. Although cetuximab in combination with paclitaxel (175 mg/m 2 ) and carboplatin (AUC 6) was well tolerated, it offered no increased benefit in terms of PFS compared with traditional cytotoxic agents alone.
Cetuximab was given as a single agent in a phase II trial of cervical cancer performed by the GOG. There were no clinical responses, and only five patients (14%) were progression free at 6 months. The combination of cetuximab with topotecan (0.75 mg/m 2 ) and cisplatin (50 mg/m 2 ) for advanced cervical cancer induced a high rate of serious adverse events, including myelosuppression, infection, and skin reaction. Furthermore, five patients died, with three deaths attributable to toxicity. Cetuximab and cisplatin (30 mg/m 2 ) for recurrent cervical cancer was better tolerated but did not show any additional benefit compared with historical rates of cisplatin alone. A recent study of single-agent cetuximab for recurrent endometrial cancer has completed accrual, and preliminary results are pending.
Trastuzumab
The overexpression of HER2 in gynecologic cancers has been associated with prognosis in endometrial cancer and other gynecologic malignancies. Trastuzumab is a humanized mAb to the extracellular domain of HER2, which prevents ligand binding. Although this drug has found great success in the treatment of Her-2/neu–positive breast cancer, its use in gynecologic oncology has been limited. Bookman and colleagues performed a phase II trial of trastuzumab in HER-2–positive recurrent ovarian cancer. Of 837 patients screened, only 11.4% were found to have high expression of HER-2. The low prevalence of this abnormality combined with a modest objective response rate of 7.3% has limited further exploration of this agent in ovarian cancer. Similarly, a phase II trial of trastuzumab (2 mg/kg) for patients with advanced or recurrent HER-2–positive endometrial cancer demonstrated no responses. Of 30 patients, 12 had SD, and the trial was closed early secondary to poor accrual. Recent data have supported the selection of only uterine serous tumors; thus, a phase II randomized trial is ongoing examining standard chemotherapy with or without trastuzumab.
Pertuzumab
The humanized mAb pertuzumab prevents the linkage of HER2 to other receptors by binding a protein site in the dimerization domain. To date, pertuzumab has only been evaluated in recurrent ovarian cancer. In a trial of two dosing regimens of pertuzumab (840 mg loading with 420 mg every 21 days vs. 1050 mg every 21 days), five patients achieved partial response, and eight patients had SD. This study found a trend toward improved median PFS and expression of phospho-HER2. The GOG evaluated pertuzumab as a monotherapy in recurrent ovarian cancer and achieved an overall response rate of 4.3%. An additional 6.8% of patients had SD for longer than 6 months. A large randomized trial comparing gemcitabine (800 mg/m 2 ) alone with gemcitabine with pertuzumab (420 mg every 21 days) found that the addition of pertuzumab resulted in improved response rates (4.6% vs. 13.8%, respectively) and a trend toward improved PFS. The greatest treatment benefit was found in patients with low HER3 mRNA expression.
Seribantumab
HER3 has also been identified as a potential target of interest in gynecologic malignancies, specifically ovarian cancer. Seribantumab is a mAb that targets HER3 to block heregulin-mediated ErbB3 signaling and chemotherapy resistance. After identification of a recommended phase II dose, seribantumab has been primarily evaluated in combination with cytotoxic and targeted therapies to treat advanced solid tumors. A randomized phase II trial of seribantumab in combination with paclitaxel revealed no difference in PFS compared with paclitaxel alone in recurrent ovarian cancer. A specific panel of biomarkers, including heregulin mRNA expression and low HER2, were associated with benefit from the addition of seribantumab to paclitaxel.
Combination Agents
Lapatinib
Lapatinib is an oral tyrosine kinase inhibitor of EGFR and HER2. It acts through binding of the ATP-binding domain and prevents pathway activation downstream. A single-agent study in recurrent ovarian cancer conducted by the GOG revealed no objective responses and only two patients progression free at 6 months (8%). As a combination, lapatinib (1000 mg/day) with metronomic carboplatin (AUC 2 weekly) and paclitaxel (60 mg/m 2 weekly) achieved response rates of 50% and SD in 30% of patients with recurrent ovarian cancer. These results were encouraging given the high number of median prior therapies in this group of patients. Other combinations with lapatinib including carboplatin and topotecan have not been as successful and have demonstrated unacceptable toxicity.
The use of lapatinib in combination with pazopanib in cervical cancer was discussed earlier in the chapter. The GOG also performed a phase II trial of lapatinib as monotherapy in recurrent or advanced endometrial cancer. Similar to the results in ovarian cancer, there was only one objective response, and seven patients achieved a best response of SD.
Multipathway Targeted Agents
Sorafenib
Sorafenib is an interesting small molecule inhibitor that effectively blocks several angiogenesis-related receptors (VEGFR, PDGFR, and KIT) and Raf, a key component of the Raf-MAPK kinase (MEK)-MAPK proliferation pathway. Sorafenib is of considerable interest in ovarian cancer that has been shown to harbor alterations in this pathway. Consequently, several trials are ongoing to evaluate the efficacy of sorafenib in the upfront, recurrent, and consolidation setting. Matei and colleagues reported the results of a phase II trial of sorafenib (400 mg bid) in recurrent ovarian cancer treated with fewer than two prior therapies. Of 73 patients, 22 achieved partial response or SD with toxicities, including rash and metabolic abnormalities. Of note, among the 59 patients with measurable disease, 12 were progression free for at least 6 months. The combination of sorafenib (200 mg twice daily) with bevacizumab (5 mg/kg) for the treatment of solid tumors was explored in a phase I study. Although the combination appeared to have significant activity in 13 patients with ovarian cancer (43% response rate), toxicity was common, leading to sorafenib dose reductions in 74% of patients. The most prevalent toxicities were diarrhea, elevated liver transaminases, hypertension, hand and foot syndrome, and fatigue.
In combination with traditional cytotoxic chemotherapy, including topotecan and gemcitabine, sorafenib has shown similar clinical benefit rates in recurrent ovarian cancer with minimal additional toxicity. A current phase III trial in newly diagnosed ovarian cancer after cytoreductive surgery seeks to determine if the addition of sorafenib (400 mg bid) to standard paclitaxel (175 mg/m 2 ) and carboplatin (AUC 6) provides any additional PFS benefit. Sorafenib is also being explored as a maintenance agent in ovarian cancer and an additional chemosensitizer in the upfront treatment of advanced cervical cancer with radiotherapy.
Vandetanib
Vandetanib inhibits the tyrosine kinase activity of two parallel pathways through action on VEGFR-2 and EGFR. This leads to suppression of angiogenesis, cell proliferation, migration, and survival mechanisms. A phase II trial of vandetanib alone to treat recurrent ovarian cancer was terminated after the first stage of accrual because no patients achieved response or SD. Of note, molecular testing in this study revealed evidence of blockage of the EGFR pathway but no activity against VEGFR. Although single-agent activity has been disappointing, this agent underwent exploration in combination with docetaxel and liposomal doxorubicin for the treatment of ovarian cancer. A randomized phase I/II study of liposomal doxorubicin alone or in combination with vandetanib was limited by significant toxicity, which led to cessation in a high number of patients treated with the combination. Conversely, in a separate randomized phase II study, vandetanib in combination with docetaxel was tolerable; however, there was no improvement in PFS compared with docetaxel alone.
Imatinib
Imatinib mesylate is an example of a small molecule that inhibits multiple pathways, including c-kit, bcr-abl, and PDGFR. Although imatinib has been quite successful for the treatment of chronic myelogenous leukemia, in which the tyrosine kinase bcr-abl is constitutively activated, the success in gynecologic malignancies as a single agent has been limited. This may result in part from the impact of the PDGF axis on pericytes, which are mesenchymal cells that support endothelial cells and newly developing blood vessels. PDGF activity recruits pericytes, which produce VEGF to stimulate angiogenesis and survival. Research on pericytes has indicated that blocking the PDGF axis in addition to the VEGF axis may be necessary to achieve improved response rates. Thus far, imatinib has only been explored as a single agent or in combination with cytotoxic chemotherapy for gynecologic malignancies.
Several phase II trials for recurrent ovarian cancer, both high and low grade, have been performed using imatinib (400 mg/day) as a single agent. Unfortunately, rates of response have been low, with only one objective responder among all trials. Rates of SD ranged from 9% to 33%. Similar disappointing results have been seen in cervical cancer. In a trial of 12 patients with recurrent cervical cancer treated with imatinib, there were no responses and only one patient with SD. In all trials, expression of drug-related targets has not been associated with response. The use of imatinib in combination with docetaxel (30 mg/m 2 weekly) in 23 patients with recurrent ovarian cancer achieved five responses, including one complete response. Three additional patients had SD lasting longer than 4 months. Further evaluations of this drug in combination with other cytotoxic chemotherapy for recurrent ovarian cancer have yielded minimal success.
Dasatinib
Dasatinib is a strong inhibitor of the Src family of kinases, which participate in the regulation of cellular growth, adhesion, invasion, and migration. At higher concentrations, dasatinib has effects on a variety of different targets relevant to tumor progression, including bcr-abl, c-kit, and PDGF. In addition, dasatinib targets the EphA2 receptor, which modulates cell migration, survival, proliferation, and angiogenesis. EphA2 overexpression correlates with poor outcome in ovarian cancer. Dasatinib (150 mg/day) has been explored in multiple solid tumors in early-phase trials with promising response rates. A phase I trial of this agent in combination with paclitaxel (175 mg/m 2 ) and carboplatin (AUC 6) was tolerable with reasonable activity. This combination has also been evaluated in advanced endometrial cancer with results pending.
Cabozantinib (XL-184)
Exploration into mechanisms of resistance to anti-VEGF therapies has suggested pathways responsive to the observed upregulation of HIF-1α. One pathway of interest is c-Met, which is also implicated in tumor growth, survival, and metastases. Cabozantinib is a small molecule inhibitor of VEGFR-2 and c-Met, which has been evaluated in a number of solid tumors including, recently, ovarian cancer. In a phase II randomized discontinuation trial, cabozantinib was administered to 68 platinum-sensitive and -resistant patients. Overall response was 24% with 35% of patients also exhibiting prolonged SD. Response rates were similar between the platinum-sensitive and -resistant cohorts. Because of better-than-expected results, the trial was terminated early. A subsequent GOG study compared this agent with weekly paclitaxel in recurrent ovarian cancer. Cabozantinib failed to meet prespecified endpoints of interest, demonstrating lower levels of response than weekly paclitaxel.
Other Targets of Interest
Angiopoietin (Ang)/Tie-2
In addition to the classic VEGF pathways of angiogenesis, other pathways involved in later stages of neovascularization hold promise for targeted therapy. Ang-1 and Ang-2 bind the receptor Tie-2 to activate pathways that stimulate endothelial cell proliferation, motility, and survival. In addition, the angiopoietins recruit pericytes to support vascular maturation. Trebananib (AMG-386) is a peptibody, a peptide fusion protein, that selectively binds Ang-1 and Ang-2, preventing interaction with Tie-2. In phase I studies of solid tumors, this compound was well tolerated, without many of the classic toxicities associated with antiangiogenic agents. A phase II study comparing two different dose levels of trebananib (3 mg/kg, 10 mg/kg) versus placebo in combination with weekly paclitaxel (80 mg/m 2 ) demonstrated a trend toward improved PFS in patients who achieved higher steady-state concentrations of trebananib. These results lead to the dose selection for TRINOVA-1, a study comparing trebananib versus placebo plus weekly paclitaxel in recurrent ovarian cancer. The addition of trebananib yielded a significant improvement in PFS period of 2 months. A phase Ib study of trebananib in combination with paclitaxel and carboplatin was found to be tolerable in women with advanced ovarian cancer. This led to development of TRINOVA-3, a randomized phase III study in upfront ovarian cancer. Results have not yet been reported. AMG-386 (10 mg/kg) has been explored in combination with liposomal doxorubicin (50 mg/m 2 ) or topotecan (4 mg/m 2 ) for recurrent ovarian cancer in a phase Ib trial. Interim results have demonstrated acceptable toxicity; however, efficacy data are not yet mature. In recurrent endometrial cancer, trebananib as a single agent did not have sufficient efficacy to pursue further evaluation, with only one objective response of 32 patients treated.
Aurora Kinase
Three members of the aurora kinase family (Aurora kinase, A, B, and C) play large roles in the phases of mitosis. These proteins are serine/threonine kinases that act to control cell division through regulation of spindle assembly, chromosome separation, and centrosome maturation. Overexpression of Aurora A and Aurora B is associated with tumorigenesis in multiple solid tumor types. Currently, there are 14 agents targeting Aurora kinase activity, which are under investigation in phase I and II trials. One agent targeting Aurora kinase A, alisertib (MLN8237), demonstrated significant activity in recurrent ovarian cancer and was evaluated as a single agent in a phase II study. Ten percent of patients treated with alisertib achieved a partial response, and 20% had SD for more than 3 months. A subsequent randomized phase II study of weekly paclitaxel alone or in combination with alisertib yielded improved PFS in the combination arm (6.7 vs. 4.7 months; HR, 0.75; 95% CI 0.58–0.96). Objective responses by RECIST were higher as well in the combination arm. An interesting new Aurora kinase A inhibitor, ENMD-2076, has additional activity against VEGFR, Flt-3, and FGFR3 kinases and has demonstrated activity in a phase I expansion cohort study of refractory ovarian cancer patients. Based on RECIST (defined later) or GCIG CA-125 response criteria, two of 20 patients and six of 20 patients, respectively, responded to single-agent therapy. This encouraged pursuit of additional phase II studies in ovarian cancer patients, which are ongoing.
Delta-Like 4 (DII4)/Notch
The Notch signaling pathway participates in the regulation of angiogenesis, proliferation, and apoptosis, making it an attractive potential target for anticancer therapy. One major avenue of Notch activation is through one of its ligands, Dll4, which creates a negative feedback loop to block cellular proliferation stimulated through VEGFR2. In addition, Notch signaling through Dll4 appears to activate cell cycle arrest through a variety of mechanisms. Notch overexpression has been demonstrated in multiple tumor types, including ovarian carcinoma. Two major therapeutic avenues exist that block signaling through the Notch pathway. The gamma secretase complex acts as a mediator of Notch signaling. Gamma secretase inhibitors are nonspecific agents that are currently in multiple phase I trials for solid tumors. Their lack of specificity has led to significant toxicity in the GI tract. Thus, antibodies that target the Dll4 ligand have been developed to reduce toxicity. These compounds have demonstrated significant activity in in vivo models and are undergoing evaluation in early-phase clinical trials. Demcizumab, an anti-Dll4 antibody, achieved reasonable dose levels with adverse events, including hypertension, fatigue, anemia, and headache. This agent is undergoing evaluation in combination with weekly paclitaxel in recurrent ovarian cancer. Other agents, including tarextumab, MK-0752, R04929097, and PF63084014, are under study in early-phase clinical trials.
Folate Receptor α
Folate receptor α binds folic acid with high affinity and is involved in folate transport. This is quite relevant in gynecologic malignancy because this receptor is overexpressed in more than 90% of ovarian cancers. Furthermore, there is minimal expression of this target in normal tissues, making this an attractive target for anticancer therapy. Farletuzumab (MORAb-003), a humanized mAb to folate receptor α, demonstrated inhibition of cellular proliferation and stimulation of cell death in preclinical models. A phase II trial of this agent (100 mg/m 2 ) in platinum-sensitive ovarian cancer was designed as a single-agent study with subsequent addition of traditional chemotherapy (carboplatin and paclitaxel) at the time of disease progression. In the single-agent arm, 24 of 28 patients remained on therapy at least 9 weeks. With combination therapy, 80% of patients achieved normalization of their CA-125, and 75% had complete or partial response. Unfortunately, farletuzumab in combination with carboplatin and paclitaxel did not yield superior outcomes compared with chemotherapy alone. Of note, a planned subset analysis found that lower pretreatment CA125 and higher exposure to farletuzumab yielded improved PFS compared with placebo.
Mirvetuximab soravtansine (IMGN853) is an antibody–drug conjugate targeting folate receptor α. The folate receptor antibody is attached to maytansinoid, a highly toxic agent that targets microtubules to induce cellular death. This agent is quite potent against cells that are folate receptor positive and even to nearby folate receptor–negative cells. A phase I trial of mirvetuximab soravtansine revealed that the agent was well tolerated, and in a small expansion cohort of ovarian cancer ( n = 17), 53% had an objective response despite platinum-resistant disease. This agent may undergo further development, alone and in combination with bevacizumab, in ovarian and endometrial cancer.
Vintafolide (EC-145)
Another agent leveraging the folate receptor α is vintafolide. This novel ligand drug conjugate combines folic acid with desacetylvinblastine hydrazide, a chemotherapeutic agent difficult to deliver systemically. In a randomized phase II trial, vintafolide combined with pegylated liposomal doxorubicin (PLD) was associated with a significantly longer PFS than PLD alone (5 vs. 2.7 months; HR, 0.63; P = 0.031). Of interest, and in support of the target selectivity, there were few differences in the observed toxicities between the two arms. Furthermore, patients with global tumor expression of folate receptor α as measured by EC-20, a tracer conjugated to folic acid, had a PFS that was similar to that of the overall population but substantially better than the control arm, suggesting that high receptor expression is associated with poor prognosis. Unfortunately, a subsequent phase III trial was stopped early because of futility, although final results have not yet been reported.
Antimesothelin Antibodies
Mesothelin is an antigen that is highly expressed in several advanced solid tumors, including ovarian cancer. Growing data indicate that this protein may have a role in the peritoneal spread of ovarian cancer. Thus, a number of antibody–drug conjugates containing an antimesothelin antibody are in development. DMOT4039A was studied in the phase I setting for unresectable pancreatic and platinum-resistant ovarian cancer. Toxicities included GI and constitutional, and many were cumulative over time. Objective activity was seen, and this agent is under further evaluation. There are several other agents in development, although no results have been reported.
Histone Acetyltransferases and Histone Deacetylases
The epigenetic modulation of gene expression is a promising target for the treatment of gynecologic malignancies. The posttranslational modification of histones is a key mechanism to regulate the expression of certain genes and has been implicated in tumorigenesis. Histones may be modified through acetylation, methylation, phosphorylation, and ubiquitination. Two enzymes, histone acetyltransferase and histone deacetylases (HDAC), mediate the addition and removal of acetyl groups, respectively. HDAC activity leads to a change in the histone structure, which leads to a change in transcription activity. Through activation of oncogenes or inactivation of tumor suppressors, cell growth, proliferation, and evasion of apoptosis may occur. HDAC inhibitors have been explored in a variety of solid tumor types. Suberoylanilide hydroxamic acid (Vorinostat) is FDA approved for the treatment of refractory cutaneous T-cell lymphoma and has been explored in recurrent ovarian cancer. A phase II study of single-agent vorinostat (400 mg/day) in 27 patients had limited success with only one partial response (4%) and two patients with SD for 6 months (7%). The HDAC inhibitor belinostat was evaluated as a single agent in platinum resistant ovary and micropapillary low malignant potential tumors. This study of 32 patients demonstrated modest activity with only one partial response and 19 patients with SD. There is evidence to suggest that the combination of HDAC inhibitors with traditional cytotoxic chemotherapy may yield improved results; thus, these agents have been explored in combination with traditional chemotherapy regimens. The combination of vorinostat with gemcitabine and carboplatin was quite toxic, although some activity was achieved. The study was terminated early secondary to overall poor tolerance. Conversely, belinostat was successfully combined with carboplatin as well as with carboplatin and paclitaxel. Although both combinations were tolerable, only belinostat in combination with carboplatin and paclitaxel yielded response rates worthy of further study.
Platelet-derived Growth Factor
In addition to the combination agents noted previously, there are agents in development that solely target the PDGFR. This is supported by recent research indicating that the v-sis oncogene is derived from the PDGF gene. Furthermore, the PDGF pathway is involved not only in angiogenesis but also in stimulation of cancer growth and regulation of fibroblasts. The use of a mAb to target this pathway promises to improve efficacy and reduce off-target effects. Olaratumab (IMG-3G3) is a mAb to PDGF-α that has demonstrated significant antitumor activity in preclinical studies in ovarian cancer cell lines. Phase I trials in advanced solid malignancy toxicity have led to the development of a current phase II study of this agent (20 mg/kg) in combination with liposomal doxorubicin (40 mg/m 2 ) for platinum-resistant recurrent ovarian cancer. As noted previously, as a result of the effects on pericytes, the use of this agent in combination with other therapies promises to improve efficacy over it as a single agent.
P53
Given its high prevalence across a number of gynecologic malignancies and advanced solid tumors, there is great interest to leverage targeted agents against p53 mutations. By far, Wee1 inhibitors are the furthest developed against this important target. Tumors with mutations in p53 are dependent on the G2 cell cycle checkpoint, which is regulated by Wee1, to repair damaged DNA. Inhibition of Wee1 creates a situation of synthetic lethality among p53 mutant tumors. AZD1775 is a Wee1 inhibitor that has been combined with carboplatin and paclitaxel in the platinum-sensitive recurrent setting of ovarian cancer. The combination of chemotherapy with a Wee1 inhibitor yielded an improved response rate and PFS compared with chemotherapy alone. This agent is now under evaluation alone and in combination with several targeted agents for ovarian cancer. In addition to Wee1 inhibitors, several molecules are in early development that appear to restore the function of p53 across a number of p53 mutant tumors in the preclinical setting.
Immune Therapy
As noted earlier, immune escape is an essential contribution to successful tumorigenesis. Over the past 5 years, the number of successful immune therapy agents has skyrocketed. Early studies have focused on notoriously immune-driven cancers, including melanoma and non–small cell lung cancer. Single-agent studies of agents targeting PD-1 and PD-L1, including avelumab, nivolumab, and pembrolizumab, have been less successful in ovarian cancer, yielding modest response rates approaching 15% in an unselected platinum-resistant setting, primarily among patients with clear cell histology. Responses among high-grade serous ovarian cancer have been limited. However, growing data indicate that activity of immune therapy may be related to common features of gynecologic malignancies, such as mutations in BRCA , POLE, or the presence of microsatellite instability. This has led to development of immune therapy trials in ovarian, endometrial, and cervical cancer both alone and in combination with chemotherapy or targeted therapy. In addition to the checkpoint inhibitor trials described earlier, the use of tumor-directed T cells (adoptive T-cell therapy) has been explored in cervical malignancies. A small study of this therapy in nine women with metastatic cervical cancer yielded three objective responses, including two durable complete responses. Certainly, these therapies are promising and results from larger clinical trials are eagerly anticipated. In addition, approaches to enhance cytotoxic T lymphocytes (CTL) entry into tumor islets will be important for improving the efficacy of immune therapies.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


