Chemotherapy has been the mainstay of systemic treatment of cancer for nearly 75 years. Chemotherapy can be broadly classified as either cytotoxic (killing rapidly dividing cells) or cytostatic (preventing further replication of rapidly dividing cells). However, chemotherapy is not specific to cancer cells, and will also affect normal cells which have high turnover, such as those in the gastrointestinal tract (leading to inflammation and sloughing of the GI tract), hair follicles (leading to alopecia), and in the bone marrow (leading to anemia, thrombocytopenia, and leukopenia). Many of the targeted therapies we use and will discuss in this chapter are cytostatic drugs, and implemented to retard growth, while traditional chemotherapies—such as alkylating agents and antimetabolites—are cytotoxic and lead to apoptosis of the affected cells.
Goodman and Gilman’s use of nitrogen mustard in the treatment of Hodgkin’s lymphoma and Farber’s use of folate analogues for acute lymphoblastic leukemia (ALL)—both cytotoxic therapies—opened the door to chemotherapy in the mid-1940s.1,2 The next step in the treatment of what were considered incurable malignancies came with the premise of combination chemotherapy, whereby several different drugs—each aimed at affecting phases in the cell cycle—were used simultaneously.3 Within a matter of months, ALL went from being an incurable disease to demonstrating prolonged remission and even cure. The concept of combinatorial chemotherapy was expanded to treat a variety of hematological and solid tumors in both children and adults. Today, combination cytotoxic chemotherapy remains the mainstay of treatment for the majority of malignancies.
Adjuvant therapy was the next major step toward our understanding of chemotherapy in the treatment of cancer with the goal to eliminate micrometastatic disease after “curative” resections. As postulated in the “cell-kill” hypothesis,4 the more abundant the tumor the less likely chemotherapy would be able to induce a cure. Fisher’s use of adjuvant therapy for breast cancer patients after radical mastectomy revolutionized the collective approach to the treatment of cancer.5 Rather than depending on “curative” resection alone, he used adjuvant chemotherapy to treat circulating tumor cells to prevent future metastases and recurrences. Since their pivotal study, and the subsequent influence of the National Surgical Adjuvant Breast Project (NSABP), an overwhelming number of clinical trials implementing combination chemotherapy have been employed in both the adjuvant and neoadjuvant setting. It was classically thought that if only the right combination of chemotherapeutic drugs for a particular malignancy could be identified, then a large-scale cure would be attainable. However, in most cases, chemotherapy is largely an extender of survival more than it is a modality of cure. Moreover, the morbidity associated with chemotherapy, as a single agent or in combination, can be debilitating, as patients often cannot tolerate the dosage or duration prescribed. More specific targeted therapy was needed.
The search for molecular targets which would allow greater precision toward tumor cells without causing damage to healthy cells began, not surprisingly, with the treatment of breast cancer. The identification of tamoxifen (a cytostatic drug) as a targeted therapy—alone or in combination with other therapy—for estrogen receptor positive (ER+) breast cancer facilitated the impetus of finding similar targets in other cancers. These targeted therapies, and in particular those related to malignancies commonly encountered by the surgical oncologist, are the focus of this chapter.
The ideal target for treatment of any particular malignancy is one that is exclusive to the cancer cell, and does not exist on healthy cells – thereby isolating the cancer without collateral damage. Such a perfect therapy remains elusive. Less ideal, but more attainable, has been the identification of a protein or molecule that is overexpressed on, or displays higher activity in cancer cells. For example, while the HER2 protein is expressed on the surface of cells throughout the body, it is overexpressed on 15% to 30% of breast cancers.6 There is an ideal target for breast cancers with a drug such as trastuzumab (Herceptin®)—a monoclonal antibody which binds to HER2 and slows cell propagation.
There are a multitude of drugs currently being used or in development, which are directed at various aspects of cancer including tumor growth, proliferation, evasion of host immunity, resistance to apoptosis or distant spread. These drugs are named according to a specific nomenclature (Table 9-1), which takes into consideration the mechanism of action, the specific target and the source from which the drug is derived.7 Herein, we discuss the most notable targeted therapies utilized in each type of tumor as they apply to the surgical oncologist.
A Generic Naming System for Monoclonal Antibodies and Inhibitory Small Molecules
| Prefix | Substem 1 | Substem 2 | Stem | |||
|---|---|---|---|---|---|---|
| Monoclonal Antibodies | (Target) | Substem | (Source) | Stem | (Type) | |
| Variable | – ci(r) – | circulatory system | – xi – | chimeric human | – mab | monoclonal antibody |
| – li(m) – | immune system | – zu – | humanized mouse | |||
| – t(u) – | tumor | – mu – | fully human | |||
| Small Molecules | (inhibitor target) | |||||
| – tin – | tyrosine kinase | – ib | small molecule with inhibitory properties | |||
| – zom – | proteasome | |||||
| – cicl – | cyclin-dependent kinase | |||||
| – par – | poly ADP-ribose polymerase | |||||
Any receptor inhibitor that targets estrogen falls into this class. While this includes many drugs used for hormone replacement therapy and contraception—which, coincidentally, is what tamoxifen was originally intended to be, we will only be focusing on SERMs’ role in breast cancer treatment. SERMs can be wholly agonistic (mimicking the action of estrogen), wholly antagonistic (competitively inhibiting estrogen), or have a mixed agonist/antagonist effect. Tamoxifenacts as a mixed agonist/antagonist, and is therefore termed a selective estrogen receptor modulator. It is the antagonist effects that relate to its function in breast cancer. It is a nonsteroidal triphenylethylene derivative that binds to, and competitively inhibits, the estrogen receptor (ER) on mammary epithelium, thereby preventing activation of ER+ cancer cells. It acts primarily on TGF-β, inciting both autocrine8 and paracrine9 effects. As an agonist, it has been shown to have a variety of functions including prevention of bone loss, and promoting endometrial hyperplasia—which has been demonstrated to increase the risk of endometrial cancer in treated patients.10 While raloxifene acts similarly to tamoxifen on breast and bone tissue, it is a benzothiophene derivative, and as such has fewer of the risks that are associated with tamoxifen. Raloxifene is also highly effective in preventing osteoporosis and acts as an antagonist on endometrial tissue, making it preferable in postmenopausal patients.
SERMs have been shown in multiple trials to reduce the risk of invasive breast cancer in both pre- and postmenopausal high-risk women.11 Current recommendations state that they should only be continued for 5 years,12 although longer use has been reported.13 For patients with a diagnosis of breast cancer, SERMs are primarily effective in ER+ or PR+ breast cancers, and it should be noted that their benefit decreases over time.14
Aromatase inhibitors (AIs) block or inactivate the aromatase enzyme, preventing the last step in the biosynthesis of estrogens: the conversion of androgens to estrogen. They are only indicated for postmenopausal women and have been shown to be more effective than SERMs in these patients.15 AIs can be divided into several classes depending on selectivity and mechanism of action, but can be broadly separated into steroidal, irreversible inhibitors (“-estanes”) and nonsteroidal, reversible inhibitors (“-ozoles”).16 As a whole there is no evidence that one AI is more effective than another.17,18 They do have a deleterious effect on bone, and osteoporosis is a valid concern in these patients. Current recommendations do not exceed 5 years of treatment. However, there are a number of trials assessing the duration and effectiveness long term.19
Human epidermal growth factor receptor 2 (HER2) is a transmembrane glycoprotein that has tyrosine kinase activity and is expressed on a variety of normal cells. However, it is overexpressed in 15% to 30% of breast cancers,6 with levels as high as 2 million receptors per cell.20 Broadly, HER2 positivity correlates with worse outcomes and poorer survival. However, the identification of targeted therapies such as trastuzumab has led to an improvement for these patients. Trastuzumab is a recombinant humanized monoclonal antibody that targets the fourth domain of the HER2 and induces antibody-dependent cellular cytotoxicity by attracting natural killer cells to the targeted cells. It also inhibits HER2-mediated cell signaling via the inhibition of the MAPK and PI3K/AKT pathways, leading to the arrest of the cell cycle.21 Lastly, it has been shown to prevent the cleavage of the extracellular domain of HER2.22 Trastuzumab has displayed efficacy in both early- and late-stage HER2+ breast cancer patients, improving overall and recurrence-free survival.23,24 There is, however, strong evidence revealing primary and secondary resistance to trastuzumab, with only modest response (11% to 26%) as monotherapy.25 It is, therefore, most effective when combined with chemotherapy in the adjuvant and neoadjuvant setting.
Gastrointestinal stromal tumors (GISTs) are sarcomas arising from the interstitial cells of Cajal. The majority of these (>80%) express the CD117 antigen, derived from the c-KIT proto-oncogene, which is abnormally activated in GIST tumors. This activated proto-oncogene leads to unregulated oncogenic cell signaling. The most common mutation occurs in exon 11, although mutations in a variety of other exons have been reported.26 Exon 11 mutations have been associated with more aggressive tumors and poorer prognosis. GISTs which lack c-KIT mutation, and are CD117 negative, most commonly arise from a mutation within another member of the tyrosine kinase family, called platelet-derived growth factor receptor α (PDGFRα). Regardless of the mutation, both these types of GISTs respond to tyrosine kinase inhibitors (TKIs) such as imatinib and sunitinib. There are a few KIT/PDGFRα negative (also known as wild-type) GISTs, some of which are associated with germline mutations in the succinyl dehydrogenase subunits (Carney–Stratakis syndrome).27 These wild-type GISTs do not respond well to TKIs, are often multicentric and are, coincidentally, less aggressive tumors following a more indolent course.
The TKIs act on the active site of the tyrosine kinase receptor. This decreases the activity of the particular kinase, while not affecting other tyrosine kinases required for cellular function. Imatinib was initially found to act on the Abelson (abl) domain of the “Philadelphia” chromosome (bcr-abl) proto-oncogene and was first formulated for use in chronic myelogenous leukemia (CML) with great success. It was subsequently determined to have effect on the tyrosine kinase activity of the c-KIT and PDGFRα proto-oncogenes as well. Its usefulness in GISTs was first noted with a complete response in a single patient with metastatic disease,28 and from there several randomized trials showed broad efficacy for these tumors as primary therapy and in the adjuvant setting.29–31 Optimal duration of adjuvant therapy with imatinib has been debated. In low-risk, completely resected GIST, current recommendations include observation or 1 year of adjuvant imatinib. In patients with intermittent to high-risk GIST (Table 9-2),32 the recommendations are for 3 years of adjuvant therapy based on the study by Joensuu et al.33 For patients who are advanced or metastatic, recommendations are for continuous therapy until tumor progression. While the side effects are generally well tolerated, the cost of these therapies has recently come under scrutiny as being prohibitive.34
For patients who progress on, or cannot tolerate, imatinib, sunitinib is the second-line therapy agent which has shown to be most effective in patients with c-KIT exon 9 or PDGFRα mutations,35 as well as acting on vascular endothelial growth factors (VEGFs), RET proto-oncogene, and many other tyrosine kinase receptors. Because of its broader targeting, its side effects are more expansive than those of imatinib. The third-line therapy for GIST is regorafenib, which was recently shown in the GRID trial to significantly improve progression-free survival in patients who no longer responded to first- or second-line therapy.36
Most colorectal cancers are sporadic and result from progressive accumulations of mutations with loss of tumor suppressor genes and activation of oncogenes. A subset of colorectal cancers (15%) can also harbor inherited (HNPCC) or acquired defects in the mismatch repair genes resulting in microsatellite instability (Table 9-3). The majority (up to 60%) of colorectal cancers display mutations in the K-ras oncogene.37 Those colorectal cancers which lack K-ras mutations, so called K-ras wild type (WT), have shown promising response to anti-EGFR-targeted therapies.
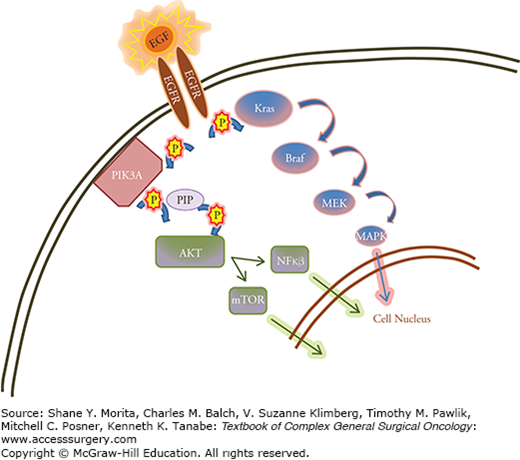
Cetuximab was the first EGFR inhibitor to be used for colorectal cancer. Its mechanism of action is on the extracellular portion of the EGFR which induces cell division via several mitogen-activated protein kinases (MAPK). When it was initially trialled, cetuximab was used as a combination therapy in patients who had failed first- and second-line therapy.38,39 Cetuximab was thought to be most useful for patients who displayed abnormal EGFR activity; however, it became obvious that EGFR expression alone was not sufficient as a marker of cetuximab efficacy. One could have predicted this based on comprehension of the MAPK pathway (Fig. 9-1), in which K-ras acts as an intracellular downstream mediator of the cell signaling pathway. A mutation in K-ras, therefore, would render upstream EGFR inhibition useless.40,41 In similar fashion, it has been shown that mutations in B-raf, the intracellular target of ras, also render anti-EGFR therapies useless.42 With improved patient selection based on these mutational status, EGFR inhibitors have shown promise as targeted therapy in combination with standard chemotherapy regimens for K-ras/B-raf WT patients, as evidenced by several trials (Table 9-4).39,43–47 Panitumumab acts in similar fashion to cetuximab, and showed modest benefit in the PRIME trial as combination first-line therapy with a platinum-based regimen.48 EGFR inhibitors as a group can cause dermatologic side effects as well as electrolyte imbalances, and patients should be periodically evaluated with lab work.
FIGURE 9-1
Diagram demonstrating epidermal growth factor receptor (EGFR)–mediated pathways to cell signaling. The epidermal growth factor (EGF) ligand attaches to the extracellular EGFR leading to dimerization and autophosphorylation of the receptor. This leads to activation of several signal transduction cascades, principally the MAPK, and AKT pathways, leading to DNA synthesis and cell proliferation.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


