This article highlights the data currently available on the activity of targeted medical treatment in a subgroup of rare entities within soft tissue sarcomas, including inflammatory myofibroblastic tumor, alveolar soft part sarcoma, solitary fibrous tumor, malignant perivascular epithelioid cell tumor (PEComa), and clear cell sarcoma.
Key points
- •
A subgroup of rare entities within a family of rare cancers was selected for responsiveness to a set of molecularly targeted agents.
- •
Low-grade tumors may respond much less to standard chemotherapy, while their higher degree of differentiation may be associated with a higher relevance of cellular pathways, which may well serve as drug-susceptible targets.
- •
Inflammatory myofibroblastic tumor carries a translocation-related target, which is strongly related to the mechanism of action of the drugs employed, while in the case of alveolar soft part sarcoma and clear cell sarcoma, the activation of MET seems to be paralleled by a lower activity of the relevant drugs.
- •
In perivascular epithelioid cell tumors, a translocation resulting in MET activation was found in a small proportion of cases, while mammalian target of rapamycin (mTOR) inhibitors were shown to have some activity, in the absence of major genetic alterations of the mTOR pathway, though in the presence of a degree of its disruption.
- •
It is possible that some of the effects seen in the clinic are caused by an unspecific mechanism of action for these targeted agents, ranging from an antiangiogenic effect to an effect on pathways that may be more or less crucial for the tumor cell.
Introduction
Inflammatory myofibroblastic tumor (IMT), alveolar soft part sarcoma (ASPS), solitary fibrous tumor (SFT), perivascular epithelioid cell tumor (PEComa), and clear cell sarcoma (CCS) are among rarer soft tissue sarcomas (STS), each of them accounting for less than 1 case per 1,000,000 population per year. They all represent translocated STS, characterized by the presence of relatively specific recurrent translocation, yet not always in the same proportion of cases. In the case of IMT and SFT, the cytogenetic aberration is responsible for the activation of 2 different proteins, anaplastic lymphoma kinase (ALK) and signal transducers and activators of transcription 6 (STAT6), respectively. Curiously, in case of ASPS, PEComa, and CCS, the 3 different histotype-specific translocations have the common feature to induce the dysregulation of the microphthalmia transcription factor (MITF) family proteins. In turn, these proteins up-regulate the transcription of MET, thus being responsible for the high metastatic rate of these sarcomas. Based on this shared molecular characteristic, ASPS, PEComa, and CCS belong to the MIT family tumors.
IMT, SFT, ASPS, PEComas, and CCS are characterized by a low (if any) sensitivity to conventional cytotoxic chemotherapy. Given the rarity of these tumors, few prospective studies focusing on their medical treatment are available. However, the well-characterized mechanisms of oncogenesis make each histotype potentially sensitive to appropriate targeted treatments. In addition, other molecular and/or morphologic peculiar characteristics (eg, the particular vascular pattern in the case of ASPS and SFT, or the activation of mammalian target of rapamycin (mTOR) in PEComas) suggest a role for new targeted agents.
This article aims to review the data currently available in the literature on the activity of targeted medical treatment in each of these histologies.
Introduction
Inflammatory myofibroblastic tumor (IMT), alveolar soft part sarcoma (ASPS), solitary fibrous tumor (SFT), perivascular epithelioid cell tumor (PEComa), and clear cell sarcoma (CCS) are among rarer soft tissue sarcomas (STS), each of them accounting for less than 1 case per 1,000,000 population per year. They all represent translocated STS, characterized by the presence of relatively specific recurrent translocation, yet not always in the same proportion of cases. In the case of IMT and SFT, the cytogenetic aberration is responsible for the activation of 2 different proteins, anaplastic lymphoma kinase (ALK) and signal transducers and activators of transcription 6 (STAT6), respectively. Curiously, in case of ASPS, PEComa, and CCS, the 3 different histotype-specific translocations have the common feature to induce the dysregulation of the microphthalmia transcription factor (MITF) family proteins. In turn, these proteins up-regulate the transcription of MET, thus being responsible for the high metastatic rate of these sarcomas. Based on this shared molecular characteristic, ASPS, PEComa, and CCS belong to the MIT family tumors.
IMT, SFT, ASPS, PEComas, and CCS are characterized by a low (if any) sensitivity to conventional cytotoxic chemotherapy. Given the rarity of these tumors, few prospective studies focusing on their medical treatment are available. However, the well-characterized mechanisms of oncogenesis make each histotype potentially sensitive to appropriate targeted treatments. In addition, other molecular and/or morphologic peculiar characteristics (eg, the particular vascular pattern in the case of ASPS and SFT, or the activation of mammalian target of rapamycin (mTOR) in PEComas) suggest a role for new targeted agents.
This article aims to review the data currently available in the literature on the activity of targeted medical treatment in each of these histologies.
Inflammatory myofibroblastic tumor
IMT is a mesenchymal spindle cell neoplasm associated with plasma cells, lymphocytes, and granulocytes in variable amount, and featuring myofibroblastic differentiation. Almost half of IMT carries a recurrent clonal aberration involving the ALK locus on chromosome 2p23. ALK is a receptor tyrosine kinase implicated in the normal development and function of the nervous system, and whatever the partner genes, the resulting chimeric protein induces a hyperactivation of the kinase activity leading to uncontrolled cell growth. IMT is a low-grade sarcoma that arises predominantly in the lung, mesentery, retroperitoneum, and pelvis of children and young adults with a propensity for local recurrences, although infrequently, IMT may metastasize. Interestingly, the more aggressive variants of IMT usually are not associated with ALK rearrangement. Surgery is the treatment of choice for localized disease; patients with unresectable IMT may benefit from steroids and chemotherapy.
Among molecular target agents, the ALK/MET-inhibitor crizotinib (Xalkori) is currently the only agent found to be potentially active in this disease.
Crizotinib is an orally available inhibitor of MET and ALK tyrosine kinases; its antiproliferative activity is derived from the competitive inhibition of the adenosine triphosphate (ATP)-binding site of both kinases. The activity of crizotinib in IMT was recently described by Butrynski and colleagues. Two patients with unresectable recurrent IMT received crizotinib within a dose escalation phase 1 trial ( NCT00585195 ). Interestingly, 1 patient, who suffered from an ALK-negative disease, progressed to the drug, while the other patient, whose disease carried the ALK-RANBP2 fusion gene, achieved sustained partial response (PR). As for tyrosine kinase inhibitor (TKI), despite initial impressive clinical activity, resistance to crizotinib occurred; after 8 months of therapy, some of the responding abdominal masses restarted to grow. The patient had debulking surgery. Genetic analyses on the surgical specimens confirmed the presence of ALK-RANBP2 fusion gene; however, sequencing analysis showed the presence of a mutation in the kinase domain of ALK, F1174L, in one of the nonresponding lesions, which was not present in the pretreatment specimen. In vitro studies demonstrated that the presence of the F1174L conferred growth advantage over the ALK-RANBP2 genotype; this was mirrored by a higher phosphorylation of ALK. Moreover, the ALK-RANBP2 F1174L conferred lower sensibility to crizotinib compared with ALK-RANBP2. Searching for a way to overcome resistance, other drugs were tested on the ALK-RANBP2 F1174L mutant; TAE684, a structurally unrelated second generation ALK inhibitor, and the HSP90 inhibitor 17AAG proved active, opening new possibilities for sequential or combination therapy. Given the clinical activity of crizotinib in patients with IMT, a European phase 2 clinical trial is actively enrolling patients with advanced disease ( NCT01524926 ).
Alveolar soft tissue sarcoma
ASPS mainly affects young patients and is composed of a distinctive epithelioid cell population organized in an alveolar growth pattern. It is characterized by an unbalanced recurrent t(X;17)(p11;q25) translocation that leads to MET transcriptional up-regulation by means of the transcription factor ASPL-TFE3. ASPS is marked by a peculiar tumor-associated vasculature and by the expression of vascular endothelial growth factor (VEGFR) and platelet derived growth factor receptor (PDGFR) on both tumor vessels and tumor cells. The natural history of ASPS is marked by an indolent behavior in spite of a greater than 60% risk of metastasis and fewer than 50% of patients disease-free at 10 years (1). ASPS is known for its resistance to conventional chemotherapy, and no standard active medical options are available for the advanced phase ( Figs. 1 and 2 ).
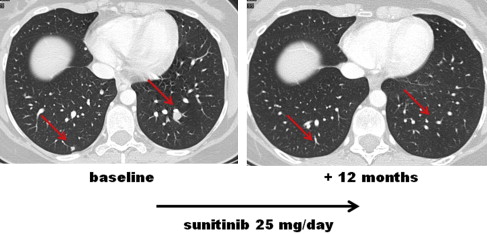
Given the molecular profile and preclinical data, MET represents a potential therapeutic target. Furthermore, the activity of antiangiogenic agents such as interferon-alfa, bevacizumab (Avastin), sunitinib (Sutent) and cediranib ( AZD2171 ) was reported, too.
In detail, a phase 2 study on the selective MET inhibitor tivantinib ( ARQ197, ) in advanced patients recently showed a modest antitumor activity of this agent in ASPS. No PR by response evaluation criteria in solid tumor (RECIST) were observed out of 27 ASPS patients who entered the study, with a 5.5-month median progression-free survival (PFS) in a slow-growing disease. Another phase 2 trial on the MET inhibitor crizotinib has just started in Europe ( NCT01524926 ), open to patients with ALK and MET-related tumors, included cases of ASPS, CCS, and IMT. No preliminary data on crizotinib in ASPS are available. Among multitargeted TKIs, the authors reported on a series of 9 patients with progressive metastatic ASPS treated with sunitinib with RECIST PR in 5 cases, and a 17-month median PFS. The sunitinib antitumor effect looked to be mediated by PDGFRB, VEGFR2, and RET and were found to be expressed on tumor vessels and also on tumor cells. A direct antitumor effect of sunitinib was also confirmed in a short-term cell culture. Following preliminary data in early phase clinical trials showing its activity in ASPS, the VEGF/KIT TKI cediranib was evaluated in a phase 2 prospective clinical study ( NCT00942877 ). The results were recently published, confirming that this agent is active in ASPS. The authors detected 35% PR plus 60% stable disease (SD) by RECIST, with a disease control rate of 84% at 24 weeks. Interestingly, most patients with a RECIST SD showed a tumor shrinkage, even if less than 30%. A randomized phase 3 study comparing sunitinib and cediranib is ongoing in the United States ( NCT01391962 ).
Solitary fibrous tumor
SFT has been initially reported as a primary mesenchymal tumor of the pleura and consequently labeled as a benign localized mesothelioma. It shows complete morphologic and genetic overlap with hemangiopericytoma (HPC), an entity that has formally abolished by the most recent World Health Organization (WHO) classification of mesenchymal neoplasm and is grouped within fibroblastic tumors. SFT can arise at almost all anatomic sites; the name HPC is still (and erroneously) retained only for tumors arising from the central nervous system (CNS). Very recently a recurrent NAB2 – STAT6 gene fusion has been detected in the vast majority of SFT, regardless of anatomic location. Interestingly the same aberration has been observed in meningeal HPCs, further proving that they represent the same entity. SFT is characterized by a broad spectrum of malignancy degree. Three clinical–pathologic variants can be schematically recognized: classical (or usual) SFT (CSFT), malignant (MSFT), and dedifferentiated (DSFT). CSFT is characterized by bland morphologic appearance and usually a favorable outcome. Unexpected recurrences with aggressive behavior can be rarely observed. MSFT is characterized by a mitotic index of at least per 10 high-power field, hypercellularity, necrosis and/or pleomorphism. DSFT’s hallmark is the presence of a sarcomatous overgrowth mimicking not otherwise specified or distinct high-grade sarcoma types. DSFT shows an increasing Ki-67 labeling and mTOR pathway activation. Other studies have emphasized the frequent expression of platelet-derived growth factor receptor (PDGFR) family members, MET, DDR1, ERBB2, and FGFR1, as well as insulin-like growth factor (IGF)2 and IGF1. Among sarcomas, SFT has the most prominent expression of IGF2. In addition, IGF2 production can be responsible for paraneoplastic hypoglycemia. SFT natural history is characterized by a high cure rate after complete surgery, with a 10% to 15% risk of metastasis. DSFT shows an aggressive behavior with a higher metastatic potential. The standard treatment of localized SFT is complete surgical resection, while medical treatment is needed in the advanced phase. Unfortunately, the expected response rate (RR) to doxorubicin-based chemotherapy is low.
Among molecular target agents, the activity of antiangiogenics such as bevacizumab in combination with temozolomide, sorafenib (Nexavar), sunitinib and pazopanib (Votrient) has been described in recent years. Responses were nondimensional in the majority of patients, and the molecular mechanisms by which the drugs inhibit tumor growth are still not well understood. The antitumor effect of IGF1R inhibitors was also reported.
In detail, bevacizumab is an anti-VEGF-A recombinant humanized monoclonal antibody. The group from MD Anderson reported on its effect in combination with temozolomide in 14 patients suffering of advanced SFT, with 14% PR and 76% SD by RECIST, for a median PFS of 10.8 months. Of interest, when Choi criteria were applied, 79% PR were classified. After the first case-reports, sunitinib activity in SFT was confirmed in a retrospective study on 31 progressive advanced patients treated at the National Cancer Institute, Milan. Best responses by RECIST were 6.5% PR and 50% SD, with 6-month median PFS. Again, a superior RR could be detected when nondimensional response evaluation criteria were applied, with 48% PR according to Choi criteria. Of interest, while PDGFRB and VEGFR2 as evaluated by immunohistochemistry were positive in all cases (PDGFRB in both tumor cells and vessels; VEGFR2 only in tumor vessels) and not predictive of response, a less aggressive morphology corresponded to an increased RR. Among antiangiogenics, pazopanib is another VEGFR1/2/3, PDGFR, and KIT inhibitor, recently approved for treatment of advanced STS. In the phase 2 study on pazopanib in STS, 3 patients suffering SFT were enrolled. Their responses were not all detailed, but at least 1 SFT patient was shown to have a computed tomography (CT) density reduction without tumor shrinkage after 3 months of therapy. No details are currently available on SFT treated with pazopanib in the recently completed phase 3 trial. Anti-IGF1R inhibitors also sound interesting in this tumor, given the molecular profile of the disease. Preliminary data on 2 patients support this hypothesis. The first report is on a dimensional response to figitumumab, a fully human immunoglobulin (Ig)G2 anti-IGF1R monoclonal antibody in an SFT patient with molecular evidence of IGF1R activation. A second patient had an impressive response to figitumumab plus everolimus (Afinitor), an mTOR inhibitor she received within a phase 1 study. A phase 2 study on the IGF1R inhibitor cituxumumab in combination with the mTOR inhibitor temsirulimus (Torisel) in STS including SFT ( NCT01016015 ) was recently published, again showing the activity of the combination in the subgroup of SFT patients.
Perivascular epithelioid cell
PEComas are rare mesenchymal neoplasms of myomelanocytic differentiation that share a distinctive cell type, descriptively named perivascular epithelioid cell (PEC). This cell, which is of unknown lineage, expresses both smooth muscle and melanocytic markers. The PEComa family includes different entities such as angiomyolipoma (AML), lymphangioleiomyomatosis (LAM), clear cell sugar tumor of the lung, and PEComa. The term PEComa practically refers to all PEComas other than AML and LAM. The clinical behavior of these diseases is extremely heterogeneous. LAM, although pathologically benign, causes cystic degeneration of the lungs, leading to progressive respiratory failure; similarly, renal AML may lead to abdominal hemorrhage or kidney failure. Epithelioid AMLs frequently display aggressive clinical behavior with local and distant recurrences. The presence of mitotic activity and necrosis correlates with aggressive behavior (malignant PEComa); however, criteria of malignancy need further validation. The evidence that PEComas may be associated with tuberous sclerosis complex (TSC), an autosomal-dominant disease caused by inactivating mutation in the tumor suppressor genes TSC1 or TSC2 , not only helped in the molecular characterization of the disease but also opened up new therapeutic opportunities. The TSC1/TSC2 complex inhibits the proliferative signals of the mTOR complex 1 (mTORC1) through inhibition of Rheb, a Guanosine-5′-triphosphate (GTP) hydrolase enzyme (ase) of the rat sarcoma family that activates mTORC1. Mutation in TSC1 or TSC2 leads to hyperactivation of the mTOR pathway. Although the molecular mechanisms responsible for the development of sporadic PEComas are only partially understood, preliminary data suggest activation of mTORC1 due to loss of TSC1 or TSC2. However, a subset of biologically distinct PEComas has been described. These PEComas, showing strong TFE3 immunoreactivity, carry rearrangements in TFE3 and have intact TSC2 , thus potentially influencing clinical history and sensitivity to therapy. Malignant PEComas arise ubiquitously. Standard treatment for patients with localized disease is wide surgery. Despite surgery, malignant PEComa may recur locally or metastasize. Anthracycline- or gemcitabine-based chemotherapy is administered in the advanced phase of disease, with unsatisfactory RR.
In addition to the randomized evidence of mTOR inhibitors’ efficacy in LAM patients, preliminary data suggest that of mTOR inhibitors can be active also in malignant PEComa.
So far, 4 different mTOR inhibitors are available for clinical purposes: rapamycin, (Rapamune) and its 3 derivatives, namely temsirolimus, everolimus, and ridaforolimus (Ariad). A randomized phase 3 placebo-controlled trial of sirolimus in patients with LAM confirmed early data; sirolimus was effective in stabilizing forced expiratory volume, improving force vital capacity and quality of life. Moreover, everolimus proved to be active in reducing the volume of subependymal giant-cell astrocytomas in a group of 28 patients with TSC, together with improvement in symptomatic patients. Afterward, Wagner and Wolff described the activity of mTOR inhibitors in single patients carrying malignant PEComa. This was followed by many other reports confirming the activity of these compounds in the disease. However, a recent retrospective analysis presented in abstract form showed responses to mTOR to be short lasting, with a 4-month median PFS. Cases of primary resistant disease were also reported. The mechanisms responsible for resistance to mTOR inhibitors in malignant PEComa are currently unknown. Based on these results, a phase 2 study on BEZ235 (Novartis, Basel, Switzerland), an orally available dual inhibitor of mTOR and PI3K inhibitors, in metastatic or unresectable malignant PEComa ( NCT01690871 ), is ongoing in Europe.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


