Melanoma is resistant to cytotoxic therapy, and treatment options for advanced disease have been limited historically. However, improved understanding of melanoma driver mutations, particularly those involving the mitogen-activated protein kinase pathway, has led to the development of targeted therapies that are effective in this previously treatment-refractory disease. In cutaneous melanomas with BRAF V600 mutations the selective RAF inhibitors, vemurafenib and dabrafenib, and the MEK inhibitor, trametinib, have demonstrated survival benefits. Early signals of efficacy have also been demonstrated with MEK inhibitors in melanomas with NRAS mutations, and KIT inhibitors offer promise in melanomas driven through activation of their target receptor.
Key points
- •
The molecular characterization of melanomas for BRAF, NRAS, and KIT mutations is increasingly essential for the optimal selection of targeted therapies and clinical trials in patients with advanced disease.
- •
In melanomas with BRAF V600 mutations, both RAF-inhibitor and MEK-inhibitor monotherapy improves overall survival. Resistance to both drug classes invariably develops but may be potentially delayed by upfront combination therapy.
- •
NRAS-mutant disease is more refractory to targeted therapy, although MEK-inhibitor monotherapy appears promising, and combination strategies are under investigation.
- •
KIT inhibitors are active in melanomas with exon-11 and exon-13 KIT mutations, with several compounds undergoing randomized phase III studies.
Introduction
Melanoma is the most deadly form of skin cancer, accounting for more than two-thirds of skin cancer–related mortality. Although most patients with localized disease can be cured with complete surgical excision, melanoma is highly malignant, and even small primary tumors have the potential to metastasize. In those who develop disseminated disease, treatment options have been limited. Melanoma has long been proved to be refractory to conventional chemotherapeutics. Dacarbazine has been considered the standard treatment for patients with metastatic melanoma for more than 30 years, yet only 5% to 15% of patients will achieve a response and no overall survival benefit has ever been demonstrated. Comparative trials of dacarbazine with other cytotoxic agents such as temozolomide, fotemustine, or platinum-based regimens, or in combination with biological agents such as interferon-α2b or high-dose interleukin-2, have failed to improve on this benchmark. During this era, the median survival for patients with metastatic melanoma was between 6 and 9 months.
Only more recently have tangible advances in the treatment of metastatic melanoma been realized. Developments in understanding of immune checkpoint regulation and the molecular biology of melanoma have laid the groundwork for 2 distinct treatment approaches that culminated in the successful phase III clinical trials of ipilimumab and vemurafenib in 2011. Ipilimumab is an antibody directed against the inhibitory cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) receptor expressed by activated T cells. Disruption of this immune checkpoint mechanism results in an enhanced T-cell–mediated antitumor response. In phase III studies, treatment with ipilimumab was associated with modest response rates and improvements in median overall survival, but a notable 10% improvement in overall survival after 2 and 3 years of follow-up. A subsequent analysis has demonstrated that this plateau in melanoma-related deaths is maintained from 3 years out to beyond 10 years after ipilimumab therapy, suggesting that a proportion of patients will develop truly durable antitumor responses. Risks associated with ipilimumab include a 15% to 20% incidence of clinically significant autoimmunity. Next-generation antibodies targeting the interaction between another negative regulator of T-cell function, programmed cell death 1 (PD-1) receptor and its ligand, appear more specific for T-cell anticancer immunity and may be associated with a higher response rate and less frequent autoimmune effects. A more thorough review of immunologic therapies in melanoma is provided elsewhere in this issue.
This article focuses on the second approach: the clinical development of specific targeted therapies in direct response to the recent discoveries characterizing common oncogenic drivers in cutaneous melanoma. In particular, mutations resulting in the constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, a key regulator of normal cellular growth and proliferation, appear to be central to the pathogenesis of most melanomas. Vemurafenib was the first of several agents now proven to target this pathway in cutaneous melanoma.
Introduction
Melanoma is the most deadly form of skin cancer, accounting for more than two-thirds of skin cancer–related mortality. Although most patients with localized disease can be cured with complete surgical excision, melanoma is highly malignant, and even small primary tumors have the potential to metastasize. In those who develop disseminated disease, treatment options have been limited. Melanoma has long been proved to be refractory to conventional chemotherapeutics. Dacarbazine has been considered the standard treatment for patients with metastatic melanoma for more than 30 years, yet only 5% to 15% of patients will achieve a response and no overall survival benefit has ever been demonstrated. Comparative trials of dacarbazine with other cytotoxic agents such as temozolomide, fotemustine, or platinum-based regimens, or in combination with biological agents such as interferon-α2b or high-dose interleukin-2, have failed to improve on this benchmark. During this era, the median survival for patients with metastatic melanoma was between 6 and 9 months.
Only more recently have tangible advances in the treatment of metastatic melanoma been realized. Developments in understanding of immune checkpoint regulation and the molecular biology of melanoma have laid the groundwork for 2 distinct treatment approaches that culminated in the successful phase III clinical trials of ipilimumab and vemurafenib in 2011. Ipilimumab is an antibody directed against the inhibitory cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) receptor expressed by activated T cells. Disruption of this immune checkpoint mechanism results in an enhanced T-cell–mediated antitumor response. In phase III studies, treatment with ipilimumab was associated with modest response rates and improvements in median overall survival, but a notable 10% improvement in overall survival after 2 and 3 years of follow-up. A subsequent analysis has demonstrated that this plateau in melanoma-related deaths is maintained from 3 years out to beyond 10 years after ipilimumab therapy, suggesting that a proportion of patients will develop truly durable antitumor responses. Risks associated with ipilimumab include a 15% to 20% incidence of clinically significant autoimmunity. Next-generation antibodies targeting the interaction between another negative regulator of T-cell function, programmed cell death 1 (PD-1) receptor and its ligand, appear more specific for T-cell anticancer immunity and may be associated with a higher response rate and less frequent autoimmune effects. A more thorough review of immunologic therapies in melanoma is provided elsewhere in this issue.
This article focuses on the second approach: the clinical development of specific targeted therapies in direct response to the recent discoveries characterizing common oncogenic drivers in cutaneous melanoma. In particular, mutations resulting in the constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, a key regulator of normal cellular growth and proliferation, appear to be central to the pathogenesis of most melanomas. Vemurafenib was the first of several agents now proven to target this pathway in cutaneous melanoma.
The mitogen-activated protein kinase pathway in cutaneous melanoma
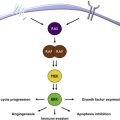
Extracellular ligands bind to specific membrane-bound receptor tyrosine kinases (RTKs) to initiate MAPK signaling. Subsequent recruitment and activation of the guanosine triphosphatase (GTPase), RAS, results in a cascade of phosphorylation (activating) events involving the serine/threonine kinases RAF, MEK, and ERK. Active ERK phosphorylates numerous cytoplasmic and nuclear targets regulating processes such as cell proliferation, differentiation, survival, migration, and angiogenesis ( Fig. 1 ).
ERK has been demonstrated to be hyperactivated in most melanomas. The most commonly identified abnormality is a mutation in BRAF , with a frequency of 40% to 60%. A single valine for glutamine substitution at codon 600 (V600E) accounts for greater than 75% of BRAF mutations, with the resultant protein having a 10-fold greater kinase activity than wild-type BRAF. NRAS mutations occur in about 15% of melanomas and are mutually exclusive to BRAF mutations. Other mutations, for instance in KIT (encoding the KIT RTK), have been identified but are far less common. However, the relative frequencies of different genetic mutations appear to cluster with certain clinicopathologic features: whereas BRAF mutations are most common in melanomas arising in skin without evidence of chronic sun damage, in much rarer mucosal or acral melanomas BRAF mutations are uncommon but the frequency of KIT mutations ranges from 10% to greater than 20%. These findings allow for more rationalized mutation testing, and have resulted in a shift from the traditional histologic classification of melanoma toward a more molecularly based one.
A further point of note is that although MAPK activation seems to be important in melanomagenesis, it is insufficient by itself to result in full malignant transformation. Evidence of this is most clearly found in the demonstration of activating BRAF mutations not only across all stages of melanoma progression but also in most benign melanocytic nevi. Other events must also be required, such as a mutation or deletion of the PTEN tumor suppressor, which co-occurs in 40% of BRAF -mutant melanomas. Nevertheless, despite this most melanomas seem to remain dependent on MAPK signaling, making this pathway an excellent therapeutic target.
Targeting BRAF in cutaneous melanoma
Early RAF-Inhibitor Studies
The earliest attempt to inhibit RAF-mediated MAPK signaling in melanoma was with sorafenib, a broad-spectrum multikinase inhibitor with pan-RAF inhibitory activity. Already in clinical development at the time of the discovery of BRAF mutations in melanoma in 2002, it was quickly adapted to be studied in patients with melanoma. Unfortunately, these studies involving sorafenib were disappointing and raised doubts as to the potential of RAF inhibition as a therapeutic strategy. In the initial single-agent phase II studies, sorafenib showed little clinical activity and no survival advantage when compared with historical controls, and although a phase I/II clinical trial of sorafenib in combination with carboplatin and paclitaxel showed promising response rates and progression-free survival (PFS) in patients unselected for BRAF genotype, no advantage for the addition of sorafenib was demonstrated in the subsequent phase III study. Preclinical experiments had shown that sorafenib could inhibit the MAPK pathway and cellular proliferation in melanoma cell lines with BRAF and NRAS mutation in vitro; however, only modest pathway inhibition was ever demonstrated in tumor biopsies from treated patients. The failure of sorafenib was therefore likely due to it being unable to achieve sufficient levels of RAF inhibition at maximum tolerated doses that were limited by its off-target multikinase activity. Subsequently, studies involving RAF inhibitors with greater potency and selectivity for mutant BRAF have followed.
Efficacy of the Selective RAF Inhibitors
Vemurafenib was the first highly selective RAF inhibitor to enter clinical trials. This agent was designed prospectively by cocrystallography of the BRAF V600E protein kinase domain and candidate compound structures. In kinase inhibition assays, vemurafenib demonstrated selective inhibition of RAF kinases at nanomolar concentrations and also demonstrated a 3-fold potency against the BRAF V600E versus BRAF wild-type kinase. In fact, in cellular assays where BRAF and CRAF signal as dimers, vemurafenib only inhibits MAPK activation in BRAF mutant cells. Surprisingly, in BRAF wild-type cells, particularly in the context of activated RAS, RAF inhibitors augmented MAPK signaling, a phenomenon now termed paradoxic MAPK pathway activation. Subsequent studies have elucidated a potential mechanism for this behavior whereby the binding of RAF inhibitors to normal CRAF and BRAF dimers induces a conformational change in these complexes that results in increased kinase activity. This differential effect of RAF inhibitors on BRAF mutant and wild-type cells has since been recognized as a major contributor to the broad therapeutic index and unique toxicity profile of this drug class.
The phase I study of vemurafenib quickly demonstrated marked differences between it and sorafenib. In the initial dose-escalation cohort, a wide therapeutic index was established, with objective tumor regressions observed at doses higher than 240 mg twice daily and dose-limiting toxicities not encountered until doses of 1120 mg twice daily. Responses were limited to patients with BRAF V600–mutant melanoma, making this population the exclusive focus of the future phase II and III vemurafenib trials. Within the subsequent expansion cohort, patients treated at the established maximum tolerated dose (MTD) of 960 mg twice daily had an unprecedented response rate of 81% and a median PFS of longer than 7 months ( Table 1 ). Paired tumor biopsies showed a strong correlation between tumor response and inhibition of cytoplasmic ERK phosphorylation. Profound inhibition of tumor metabolic activity was also demonstrated on day-15 [ 18 F]fluorodeoxyglucose positron emission tomography (FDG-PET) in all patients treated above the drug threshold of 240 mg twice daily. Similarly impressive results were reported in the phase II study, which involved 132 patients with previously treated melanoma. The confirmed objective response rate was 53%, although most patients obtained some degree of tumor control. The median PFS was 6.8 months, and the median overall survival of 15.9 months far exceeded benchmarks set in previous second-line studies.
| Study/Treatment Group | BRAF Genotype | Patients (n) | ORR (%) | Median PFS (mo) | Median OS (mo) |
|---|---|---|---|---|---|
| Phase I study of vemurafenib: dose escalation and cohort extension in previously treated patients | |||||
| Vemurafenib >240 mg bid | V600E | 16 | 69 | NR | NR |
| Vemurafenib 960 mg bid | V600E | 32 | 81 | >7.0 | NR |
| Phase II study of vemurafenib in previously treated patients | |||||
| Vemurafenib 960 mg bid | V600E | 132 | 53 | 6.8 | 15.9 |
| Phase III study of vemurafenib vs dacarbazine in untreated patients | |||||
| Vemurafenib 960 mg bid | V600E | 295 | 59 | 6.9 | 13.3 |
| V600K | 33 | 45 | 5.9 | 14.5 | |
| DTIC 1000 mg/m 2 every 21 d | V600E/K | 338 | 8.6 | 1.6 | 9.7 |
| Phase I study of dabrafenib: dose escalation and cohort extension in previously treated patients | |||||
| Dabrafenib 35–300 mg bid | V600E | 27 | 78 | 5.5 | NR |
| V600K | 18 | 39 | 5.6 | NR | |
| Phase II study of dabrafenib in previously treated patients | |||||
| Dabrafenib 150 mg bid | V600E | 76 | 60 | 6.3 | 13.1 |
| V600K | 16 | 13 | 4.5 | 12.9 | |
| Phase III study of dabrafenib vs dacarbazine in untreated patients | |||||
| Dabrafenib 150 mg bid | V600E | 187 | 53 | 5.1 | NA |
| DTIC 1000 mg/m 2 every 21 d | V600E | 63 | 6 | 2.7 | NA |
Confirmation of improved survival in patients treated with vemurafenib would come with the phase III clinical trial. This study randomly assigned 675 patients with untreated advanced melanoma harboring BRAF V600E mutations between either single-agent vemurafenib or dacarbazine. Overall survival was the primary end point, and crossover to vemurafenib was not initially permitted. At the time of the first interim analysis, with a median follow-up of only 3.7 months, the survival outcomes were clearly in favor of patients being treated with vemurafenib. Relative risk of death was reduced by 63% and of tumor progression by 74%. These results led to the early termination of the trial, and dacarbazine-treated patients were allowed to cross over to vemurafenib. Later analysis at a median follow-up of 10.5 months showed a median overall survival in vemurafenib-treated patients of 13.6 months, and 9.7 months for patients treated with dacarbazine. Objective response rates were 62.6% versus 9.8% in favor of vemurafenib. Based on these findings, vemurafenib was approved for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutations by the US Food and Drug Administration in August 2011; and in melanomas with any BRAF V600 mutation by the European Medicine Agency in February 2012.
The successes of vemurafenib were not isolated events in the development of highly targeted, mutation-specific therapeutics for melanoma. Clinical trials of other selective RAF inhibitors quickly followed. Chief among these were the trials involving dabrafenib, another inhibitor of RAF kinase with selectivity for mutant BRAF in kinase panel screening. In corresponding phase I, II, and III studies, dabrafenib showed properties similar to those of vemurafenib (see Table 1 ). Once again a high therapeutic index was apparent, and investigators had difficulties defining an MTD. Eventually a recommended phase II dose of 150 mg twice daily was brought forward, as beyond this there was little improvement in clinical responses, and near maximal effects were already noted on the pharmacodynamic studies that included tumor biopsies and FDG-PET imaging. At this dose confirmed response rates were consistently higher than 50%, and in the phase III study comparing treatment with dabrafenib versus dacarbazine in patients with untreated BRAF V600E–positive melanoma, risk of tumor progression was once again reduced by more than 70%. The median overall survival also favored patients treated with dabrafenib (hazard ratio 0.76, 95% confidence interval 0.48–1.21) but was not statistically significant, with the 36 of 63 (57%) dacarbazine-treated patients crossing over to dabrafenib and potentially obscuring any overall survival benefit obtained with initial treatment. The key differences between the vemurafenib and dabrafenib clinical trials were the intentional inclusion of patients with non– BRAF V600E mutations and patients with untreated brain metastases in the phase I and II studies of the latter.
RAF Inhibitors in BRAF Non-V600E Mutant Melanoma
Although most BRAF -mutant melanomas are the result of a V600E substitution, approximately 5% to 10% are V600K, 5% V600R or V600D, and a minority involves non-V600 abnormalities. Cellular assays suggest vemurafenib to be active against all 3 of these non-V600E codon 600 BRAF mutations, although its clinical trials used a sequencing method that was unable to reliably distinguish between V600E mutations and these variants. However, subsequent Sanger sequencing and pyrosequencing identified 33 patients with V600K mutations from the vemurafenib treatment arm of the phase III study. These patients had a response rate (45%) and survival outcome (see Table 1 ) similar to those with V600E mutations. The phase I and II trials of dabrafenib intentionally enrolled patients with non-V600E mutations and included a total of 67 patients with V600K mutations, including 34 patients with untreated brain metastases. Individual trials were not sufficiently powered to detect significant differences between V600K- and V600E-bearing melanomas, and although clear benefits were seen in both, response rates and survival outcomes were slightly inferior in the V600K populations. As the phase III study enrolled only those patients with confirmed V600E mutations, more detailed information is not available. However, like vemurafenib, dabrafenib has similar activity against V600E and V600K mutant BRAF in kinase assays, and on the balance of current evidence it is believed that most patients with codon 600 BRAF mutations are likely to benefit from RAF-inhibitor therapy. For patients with noncodon 600 BRAF mutations the data are even more limited, although it seems unlikely that these patients benefit. Cell lines expressing BRAF K601E require significantly higher concentrations of vemurafenib to suppress growth that may be difficult to achieve in vivo, and in clinical trials there were no responders among the 3 patients with non-V600 BRAF mutations (2 K601E and 1 V600_K601delinsE mutation) treated in the phase I study of dabrafenib.
RAF Inhibitors in Patients with Brain Metastases
Brain metastases are present at diagnosis in up to 20% of patients with metastatic melanoma, and develop in 40% to 45% of patients overall. Treatment options have included surgery, stereotactic radiosurgery, or whole brain irradiation. However, the response rate to systemic chemotherapy is less than 10%, and their presence is a poor prognostic feature, signifying a median overall survival of only 4 to 5 months.
Because of concerns about the potential for neurotoxicity, dabrafenib was specifically engineered not to cross the blood-brain barrier. Investigators were therefore surprised by evidence of intracranial activity in a small group of patients with brain metastases enrolled in the phase I trial’s expansion cohort. These findings resulted in a subsequently designed phase II study that enrolled 172 patients with either untreated or secondarily progressive melanoma brain metastases. In the subset of 139 (81%) patients with V600E mutations, the intracranial response rate was 39.2% in those with untreated brain metastases and 30.8% in those with previously treated brain metastases. Median PFS was just over 16 weeks and the median overall survival just over 31 weeks. Similar to in extracranial disease, slightly less robust results were noted in the 33 patients with BRAF V600K mutations. The results suggested superiority to previous systemic agents in patients with intracranial metastases, and confirmed the activity of dabrafenib in this setting.
Based on its physiochemical properties, vemurafenib was also expected to have difficulty crossing the blood-brain barrier, and patients with cerebral disease were specifically excluded from its initial clinical trials. However, a subsequent phase II open-label study in 24 patients with cerebral disease demonstrated similar PFS benefits to patients treated with dabrafenib, and a second larger trial is currently under way to further define its efficacy. The explanation underlying the activity of these RAF inhibitors in central nervous system disease is probably related to the disruption of the normal blood-brain barrier integrity in the presence of melanoma macrometastases. These agents now provide a viable option for the treatment of patients with cerebral disease, and should encourage the inclusion of similar patients in future studies of targeted therapies in melanoma.
RAF-Inhibitor Toxicity
RAF inhibitors as a class have been generally well tolerated. In clinical trials approximately 25% of patients required a dose interruption or modification, and 5% drug cessation. Common toxicities include arthralgia, fatigue, headache, and nausea. Particular to vemurafenib is photosensitivity, rare cases of prolonged QT interval, uveitis, and bilateral facial palsies. Febrile reactions are frequent in patients treated with dabrafenib.
Also common to both drugs are prominent cutaneous side effects, which have included rash, hyperkeratosis, and papillomas. However, a particular concern has been the development of either cutaneous squamous cell carcinoma and keratoacanthomas in 20% to 26% and 6% to 11% of patients enrolled in the phase I to III studies of vemurafenib or dabrafenib, respectively. Molecular studies have demonstrated that these squamous cell tumors are caused by the paradoxic activation of the MAPK pathway under the influence of RAF-inhibition. A high percentage of excised lesions have been identified as possessing RAS mutations which, combined with the short latency period in their development, have supported a drug interaction with preexisting RAS mutations in the skin. The combination treatment of a RAF inhibitor and MEK inhibitor attenuates this paradoxic signaling, reducing the incidence of cutaneous tumors. In fact, a reduced incidence of RAF-inhibitor–related rash, hyperkeratosis, and papilloma with BRAF/MEK combination therapy suggests that these are also mediated by paradoxic MAPK pathway activation.
Cases of new nevi and second primary melanomas have also been reported. Genotyping of these lesions have demonstrated them to be consistently BRAF wild-type, with one genotyped melanoma so far identified with an NRAS mutation. The true incidence (which is probably less than 2%) and significance of these lesions is yet to be determined. There have also been 2 cases of patients developing new second malignancies reported while receiving RAF-inhibitor therapy. Both of these cases were likely related to paradoxic MAPK signaling promoting the growth and expansion of a preexisting, but subclinical, RAS -mutant cell population. In the context of metastatic melanoma, this low incidence of secondary malignancies is easy to accept. However, caution will be required in the future use of these agents, particularly in the adjuvant setting, or in patients with a history of RAS -mutant cancer.
Development of RAF-Inhibitor Resistance
Although most patients will benefit from RAF-inhibitor monotherapy, approximately 10% have primary resistance and half will develop secondary resistance within a median time of 5 to 7 months. Postprogression biopsies show reactivation of the MAPK pathway in most patients progressing on vemurafenib. Underlying mechanisms so far identified include secondary mutations in NRAS ; amplification of BRAF ; expression of BRAF splice variants that form active BRAF dimers ; overexpression of the cancer Osaka thyroid (COT) kinase (an alternative MEK activator) ; or mutations in MEK itself. In patient biopsies with persistent inhibition of MAPK, the activation of RTKs including PDGFR, IGF1R, FGFR3 and MET, or loss of PTEN expression, can activate the PI3K pathway and confer RAF-inhibitor resistance. A more detailed review of resistance mechanism to RAF-inhibitor therapy is available elsewhere in this issue; however, it is clear that further strategies will be required to optimize survival in BRAF -mutant melanoma.
MEK-Inhibitor Monotherapy and Combination Therapy
As the major substrate of RAF kinase, MEK offers an alternative target for both treatment-naïve BRAF -mutant melanoma and for melanomas that develop resistance through the reactivation of the MAPK pathway. In preclinical studies, BRAF -mutant cell lines and melanoma xenografts are extremely sensitive to MEK inhibition. However, clinical trials involving early MEK inhibitors were associated with low response rates, presumably as these agents were unable to achieve sufficiently sustained target inhibition. Since then, several MEK inhibitors with more favorable pharmacokinetic and pharmacodynamic properties have been developed. Chief among them has been trametinib, which in single-agent studies has consistently shown a greater than 20% confirmed response rate ; moreover, in a phase III study, trametinib significantly improved both PFS and overall survival when compared with treatment with systemic chemotherapy ( Table 2 ). Across its phase I/II studies, trametinib demonstrated a greater breadth of activity than did the selective RAF inhibitors. Responses were seen in 3 of 4 noncodon 600 BRAF -mutant melanomas, including 2 with K601E and 1 with a L597V mutation; as well as prolonged stable disease in 1 patient with an NRAS -mutant disease. Minimal clinical benefit was obtained in the cohort of 40 patients previously treated with RAF inhibitors, suggesting a high cross-selectivity for both RAF-inhibitor and MEK-inhibitor resistance, perhaps involving a switch in dependence to alternative signaling pathways such as the PI3K pathway. Consistent with prior MEK-inhibitor studies, the common and dose-limiting toxicities included diarrhea, acneiform rash, and ocular toxicity including a reversible central serous retinopathy.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


