Fig. 12.1
Mediators of tumor angiogenesis
Antiangiogenic Agents
Bevacizumab
Bevacizumab is a recombinant, humanized, monoclonal immunoglobulin G1 antibody targeted at VEGF-A, which is approved for the treatment of metastatic colorectal cancer, non-squamous non-small cell lung cancer, glioblastoma, and metastatic renal cell carcinoma [12]. This antibody binds to and neutralizes all forms of VEGF-A, which suppresses growth of tumors and inhibits progression of metastatic disease [13, 14]. Also, anti-VEGF drugs enhance the effects of chemotherapeutic agents by improving the structure and function of tumor vessels [15]. Two phase II studies of bevacizumab monotherapy conducted on patients with recurrent ovarian or primary peritoneal cancers found response rates of 15.9–21.0 % [16, 17]. Also in one of those studies, gastrointestinal perforation (GIP) was observed in 5 of 44 (11.4 %) patients who had been treated with three or more regimens [17]. Bevacizumab was evaluated combined with first-line chemotherapy (paclitaxel + carboplatin) for ovarian cancer [18, 19]. Two phase III studies, the Gynecologic Oncology Group (GOG) 218 and the International Collaborative Group for Ovarian Neoplasia (ICON) 7, examined the combined effects with bevacizumab and standard chemotherapy (paclitaxel + carboplatin) in a first-line/adjuvant chemotherapy setting and results are shown in (Table 12.1) [20, 21]. The Ovarian Cancer Education Awareness Network (OCEANS) trial evaluated the efficacy of bevacizumab in combination with carboplatin and gemcitabine in patients with recurrent, platinum-sensitive ovarian, primary peritoneal, or fallopian tube cancers after frontline, platinum-based therapy [22]. In this study, PFS for the bevacizumab arm was significantly longer than for the placebo arm (median PFS, 12.4 vs. 8.4 months). Grade 3 or higher hypertension and proteinuria occurred more frequently in the bevacizumab arm. A multicenter, open-label, randomized, two-arm, phase III trial, AURELIA, was conducted on patients with ovarian, primary peritoneal, or fallopian tube cancers resistant to platinum [23]. It was observed in this study that chemotherapy plus bevacizumab significantly improved PFS (median PFS, 6.7 vs. 3.4 months) and overall response rate (30.9 vs. 12.6 %) compared to chemotherapy alone. Hypertension ≥ grade 2 (11.1 vs. 3.9 %) and proteinuria (6.4 vs. 0.5 %) occurred more frequently in the bevacizumab arm. GIP was observed in 1 % patients in the bevacizumab arm.
Table 12.1
Phase III trials of bevacizumab in ovarian cancer
Trial | Patients | Treatment | Median PFS (M) | Median OS (M) | Selected adverse events | Reference |
|---|---|---|---|---|---|---|
First line | ||||||
GOG 218 | 1873 | [20] | ||||
Arm 1 | 625 | CP | 10.3 | 39.3 | 1.2 % GI events (G ≥2), 7.2 % HT (G ≥2), 5.8 % VTE (any grade) | |
Arm 2 | 625 | CP + Bev | 11.2 | 38.7 | 2.8 % GI events (G ≥2), 16.5 % HT (G ≥2)***, 5.3 % VTE (any grade) | |
Arm 3 | 623 | CP + Bev → Bev | 14.1* | 39.7 | 2.6 % GI events (G ≥2), 22.9 % HT (G ≥2)***, 6.7 % VTE (any grade) | |
ICON7 | 1528 | [21] | ||||
Arm 1 | 764 | CP | 17.4 | 1.3 % GI events, 0.3 % HT, 1.7 % VTE | ||
Arm 2 | 764 | CP + Bev → Bev | 19.8* | 2.1 % GI events, 6.2 % HT, 4.3 % VTE | ||
Second line | ||||||
OCEANS | 484 | [22] | ||||
Arm 1 | 242 | CG | 8.4 | 35.2 | 0.4 % HT, 0.9 % proteinuria, 2.6 % VTE | |
Arm 2 | 242 | CG + Bev | 12.4* | 33.3 | 17.4 % HT***, 8.5 % proteinuria***, 4.0 VTE | |
AURELIA | 361 | [23] | ||||
Arm 1 | 182 | Chemotherapy alone | 12.6 | 2.2 % TEE | ||
Arm 2 | 179 | Chemotherapy + Bev | 30.9** | 2.8 % TEE | ||
Cediranib
Cediranib is a potent inhibitor of tyrosine kinase of VEGFR-1, VEGFR-2, and VEGFR-3 and c-Kit that competes for the ATP-binding site within the receptor kinase domain [24, 25]. In a phase II studies, patients with recurrent ovarian, fallopian tube, or peritoneal cancers were treated with cediranib, and 17.4 % of patients showed a partial response [26]. Grade 3 hypertension was observed in 46 % of patients. No GIP or fistulas were observed.
Pazopanib
Pazopanib inhibits tyrosine kinase of VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR-α, PDGFR-β, and c-Kit. It has been approved for renal cell carcinoma and soft tissue sarcoma [27]. A CA-125 response was observed in 31 % of patients with median response duration of 113 days during a phase II trial of pazopanib for the treatment of recurrent ovarian, fallopian tube, and primary peritoneal carcinomas [28]. The most common adverse events, noted in this trial, were elevated grade 3 alanine aminotransferase and γ-glutamyl transpeptidase, fatigue, and diarrhea.
Classical Transgene Delivery
Over the past several years, there has been an increased understanding of the mechanisms behind transgene delivery. Efficient full gene (s) delivery to a mammalian cell involves several steps: delivery of genes to target cells, cellular entry, endosome escape, and integration into the nucleus [28, 29]. No single gene transfer system has been developed that can effectively fulfill the requirements for all of the features noted above. A combination of multifunctional nonviral gene transfer systems may satisfy the requirements for efficient gene delivery. Cationic gene carriers can facilitate the cellular entry of DNA by interacting with the negatively charged cell surface [29, 30]. Endosomal escape is achieved by including components that induce membrane fusion at low pH or block pH lowering inside the endosome in the DNA-carrier complex. PEI is a cationic polymer that serves as an effective vehicle for in vivo gene delivery in many tissues. It facilitates effective DNA binding and protection, combined with the capability to escape the endosome due to its proton buffering ability with resultant osmotic swelling and endosomal disruption [30]. PEI has been shown to be an efficient transfection agent for ovarian carcinoma cells by intraperitoneal injection [31]. Several studies indicated that PEI-mediated transgene expression was dose dependent and transient, with maximal transgene expression observed during 48 h immediately following transfection [32]. DNA was detectable in very small amounts after 72 h, indicating probable degradation and clearance [31]. This would indicate that development of nonviral gene transfer technologies that can support chromosomal integration and persistent gene expression in vivo is desirable. Transposon-mediated DNA delivery systems have shown to allow the development of a new generation of vectors for human gene therapy and mammalian [33]. Transposon-based systems have been proven to be effective integrating nonviral vectors that can mediate long-term in vivo transgene expression [34]. It has been shown that SB transposons are an efficient intratumoral gene transfer vector for glioblastoma multiforme and was delivered using PEI as gene transfer reagent. The investigators of these two studies observed a marked antitumor activity as reflected by reduced tumor vessel density, inhibition of tumor growth, and tumor elimination in up to 50 % of nude mice [35, 36]. In other studies, a more active transposon used [37–42], combined with PEI to deliver HSV-TK gene into ovarian cancer xenografts. Further studies are needed to determine how PB transposon performs in vivo in the long term. In addition, the potential safety issues from insertional disruption/deregulation in genome needed to be addressed.
DNA Vectors
Adenoviruses are well-understood vectors for gene therapy. Human adenovirus serotype 5 (Ad5) is a reasonable vector to use, as it is not associated with a serious disease [43]. It has several advantages: it is well characterized, it is not restricted to infecting only dividing cells, and it can be grown in high titers with relative biologic stability allowing it to be administered systemically. Early clinical trials utilizing no replicative adenoviruses demonstrated the feasibility and potential utility of adenoviral-based gene therapy strategies for the treatment of malignancy. While the initial adenoviral-based trials utilized no replicative vectors, subsequent studies investigated the potential of conditionally replicative adenoviruses (CRAd) for the treatment of cancer. These viral agents are designed to infect cancer cells specifically, replicate selectively within these cells, and then to spread laterally to other cancer cells. A key element of CRAd development is to replicate in cancer cells selectively, without damaging normal host cells. CRAds have thus been engineered to selectively replicate within tumor cells using two broad strategies. The first employs deletion of part of the E1A or E1B genome to prevent replication in normal cells but allow replication in tumor cells with genetic defects that complement the deleted viral genome functions. The second employs a tumor-selective promoter (TSP) to drive the expression of genes involved in viral replication selectively in tumor cells. An example of the first strategy is the E1B-negative adenovirus (dl1520), Onyx-015 [44–51]. In this approach, the E1B–55 k gene of Onyx-015 was deleted, resulting in a virus that would replicate selectively within p53-deficient tumor cells. These viruses expressed the E1A protein, which overrode the block imposed by p53 in infected cells, resulting in apoptosis of normal cells, in addition to tumor cells. ONYX-015 demonstrated notable results in both in vitro and in vivo studies but had limited effects in human clinical trials for ovarian cancer. Bischoff et al. demonstrated that ONYX-015 was capable of efficient, selective replication in p53-deficient human tumor cells [52]. Heise et al. demonstrated in vivo correlation in three nude mouse–human ovarian carcinomatosis xenograft models [53]. Clinical studies of patients with head and neck cancer demonstrated safety and efficacy, but Vasey et al. failed to demonstrate similar results in ovarian cancer patients. Fifteen of the 16 study patients were taken off of the study because they developed progressive disease [54].
Enhancing Vector Infectivity
The major limitation of gene-based therapies lies within the vector systems. As the majority of patients have been previously exposed to Ad5, there is high level of immunity to the virus with preformed antibodies. In addition, primary tumor cells have low levels of the coxsackie and adenovirus receptor (CAR) relative to their cell line counterparts [55–57]. For the adenovirus vector to facilitate expression of the therapeutic gene in target cells, three steps must occur: first, the adenovirus must attach to the above receptors, second it must be internalized into the cell, and third it has to transfer the gene to the nucleus where it can be expressed. The adenovirus binds to the primary high-affinity CAR receptor and to the integrins αυβ3 and αυβ5 using two capsid proteins, the fiber and penton base, respectively. The carboxy-terminal (C-terminal) knob domain of the adenovirus fiber protein is the ligand for attachment to CAR [58–60]. It has been demonstrated that adenovirus infection is dependent upon the C-terminal knob [61]. After attachment the adenovirus is internalized using receptor-mediated endocytosis. This is reliant upon the interaction of the Arg–Gly–Asp (RGD) sequences in the penton base with the integrins αυβ3 and αυβ5 [62]. After internalization, the virus enters the cytosol via a pH-induced conformational change. The virus then localizes to the nuclear pore and is translocated into the nucleus of the host cell. One method of improving adenoviral tropism in low CAR-expressing cells such as ovarian cancer cells is by replacing the serotype knob in order to utilize a different receptor other than CAR. It has been shown that the knob domain of serotype 3 binds to CD46 and possibly CD80 and CD86, instead of CAR [63–65]. The 5/3 mutated adenovirus incorporates the alternate fiber knob domain from serotype 3 increasing the cytopathicity of a CRAd [66]. In another study, Ad5/3 has been shown to enhance tumor cell destruction in an orthotopic murine model of ovarian cancer [67] by a Δ24 CRAd with the Ad5/3 fiber chimera. Another method to improve transduction of the adenovirus is by utilizing fiber peptide modifications [67]. Ad fiber protein can enhance transduction in ovarian cancer cells by overcoming the low CAR expression. However, this method has been limited by the fact that the addition of more than 27 amino acids or more to the C-terminal end of the fiber will inhibit trimer formation [68]. X-ray crystallography showed that the HI loop is exposed on the fiber knob of the adenovirus and will allow the addition of up to 83 amino acids without affecting replication [69]. Incorporation of the RGD motif into the HI loop as compared to the C-terminus exhibited an increased level of gene transfer in in vitro studies [70, 71], and it also retargeted viruses to ovarian cancer cells that were previously resistant to adenovirus infection [56]. These tropism modifications have been incorporated into the context of CRAds, and infectivity enhancements have been achieved [72].
Achieving Selectivity with Tumor-Specific Promoters
The use of tumor-specific promoters (TSPs) is a strategy to drive viral E1 expression increasing tumor specificity. This strategy showed improved efficacy and tumor specificity [73]. TSPs have also been incorporated into CRAds. This CRAd demonstrated efficient replication and oncolysis in vitro and therapeutic efficacy in murine models of ovarian cancer [73]. Other TSP examples include CXCR4 and survivin. CXCR4 is a chemokine receptor gene that plays a role in progression and metastasis in ovarian cancer cells. A CRAd modified to include a CXCR4 TSP (CRAd–CXCR4.RGD) has demonstrated improved viral infectivity and tumor specificity in both established ovarian cancer cell lines and in primary ovarian cancer tissue samples [49]. Survivin is a protein that inhibits apoptosis, allowing the survival and progression of cancer cells [74]. In addition, CRAd showed a decreased hepatic uptake [75].
Armed CRAds
CRAds killing ability can be enhanced by constructing an “armed” entity [76]. Several aspects must be considered when constructing an “armed” CRAd. The adenoviral genome can only encapsulate approximately 105 % of the wild genome, or 38 kb; therefore the size of any insert is limited [77]. In addition, the efficacy of the virus is dependent upon replication, and the insertion of a transgene should not interfere with this critical feature. CRAds can be armed with a variety of transgenes that directly enhance the killing of the cancer cell. The most commonly used transgenes that affect cell killing are the “suicide genes” that encode prodrug-converting enzymes that convert nontoxic prodrugs into toxic metabolites. These metabolites then diffuse away from the expressing cell and kill neighboring cells as a “bystander effect.” An early example of this system is the herpes simplex virus thymidine kinase/ganciclovir (HSV-TK/GCV), which replaces the E1B–55 k gene with HSV-tk. This system is very dependent on the E1B–55 k gene as well as the amount of ganciclovir given. Another system is CD-5-FC that converts prodrug 5-FC into 5-fluorouracil (5-FU) which can be incorporated into both RNA and DNA, making it toxic to both dividing and nondividing cells [78]. CRAds constructed with short hairpin RNAs directed against vascular endothelial growth factor (VEGF) and/or interleukin 8 have also demonstrated improved antitumor efficacy [56, 57]. Immunomodulatory transgenes are designed to stimulate the immune system into recruiting immune cells to the site of the tumor and to enhance the cells’ proliferation and activation. Examples of immunomodulatory CRAds include CRAds armed with TNFα, IFN-γ, and IL-4 [79–84].
Incorporating Imaging into CRAds
An ideal noninvasive monitoring system would emit a signal that directly correlated with the level of viral replication, would be associated with the packaging of the virions to allow detection, and would be continued in viral progeny. To be effective, the system would have to minimally disrupt the replication and spread of the virus. An early approach at noninvasive imaging included positron emission tomography (PET)-directed thymidine kinase as a reporter of oncolytic herpes simplex replication [85]. There are three broad categories of reporters used to monitor the systemic distribution of CRAds in vivo. The first category consists of secreted reporters that are soluble proteins not existent in low levels in serum. [86]. The second category of reporters allows the cells to be directly visualized by arming the CRAds with imaging molecules [64–67]. A third system consists of reporters that have been incorporated into the viral capsid. This allows visualization of the viral particle itself [68]. For example, the expression of human somatostatin receptor subtype 2 (SSTR) can be followed by intravenous administration of radiolabeled somatostatin [87].
Cellular Delivery of CRAds
Therapeutic agents often have decreased efficacy in vivo compared to in vitro studies due to physiologic variants. The accurate delivery of these agents to the target tissue can enhance their therapeutic efficiency and decrease toxicity. There are several delivery systems including liposomes, microspheres, synthetic polymers, and protein DNA complexes [88, 89]. Human mesenchymal stem cells have been utilized to improve the delivery of an Ad 5/3 CXCR4 CRAd to pulmonary metastasis in a murine model of advanced breast cancer [90].
Immunotherapy Therapy for Ovarian Cancer
The available scientific data suggest that immunotherapy for ovarian cancer (OC) could be effective. Ovarian cancer cells express tumor-associated antigens against which specific immune responses have been detected. The tumor immune surveillance plays an important role in clinical outcomes in OC, as manifested by the correlation between survival and tumor infiltration with CD3+ T cells.
Animal studies and human clinical trials have demonstrated that therapeutically induced tumor-specific immunity is an efficient methodology to treat human cancer based on facts that support this concept. Tumors generally can prevent tumor-specific immunity from eliminating tumors by inducing tumor-specific tolerance or inflammation. The effective immunotherapy for ovarian cancer will have to surmount these obstacles for optimal effectiveness.
Cancer Vaccines
Vaccines have been the main approach to ovarian cancer immunotherapy, as with many other types [91–94]. Vaccines have limited efficacy as monotherapy in patients with advanced and recurrent ovarian cancer. Some studies’ results provide encouragement for vaccine development and optimization for use. Sabbatini et al. [95] noted that patients vaccinated with monovalent or heptavalent vaccines against carbohydrate epitopes experienced significantly longer time to progression and higher progression-free survival rates. Also, vaccination with anti-idiotype ACA-125 resulted in CA-125-specific antibodies and was associated with prolonged survival [96]. Carcinoembryonic antigen–MUC-1–TRICOM poxviral-based vaccines were used in 16 patients, including three of them being ovarian cancer patients. Immune responses to MUC-1 and/or carcinoembryonic antigen were seen after vaccination in nine patients. A patient with clear-cell ovarian cancer and symptomatic ascites had a radiographically and biochemically clinical response [97]. The limitation in vaccine development in ovarian cancer is due to the lack of well-characterized rejection antigens and by notable molecular heterogeneity of the disease. HER2 is a rejection antigen, and it may also serve as a rejection antigen in ovarian cancer [98, 99]. Vaccination against HER2 has resulted in sustained antigen-specific T cell, humoral immunity, and epitope spreading in patients with ovarian cancer [100]. NY-ESO-1 is a bona fide ovarian cancer rejection antigen, and it is expressed in less than 30 % of ovarian cancers [101, 102]. A viable alternative to vaccines are whole-tumor antigen vaccines created using tumor cells, autologous tumor lysate, or tumor-derived RNA [103–105]. The advantages of these vaccines are the opportunity to induce immunity to a personalized and broad range of antigens, which could minimize the development of tumor escape variants; the inclusion of yet unidentified tumor rejection antigens; no HLA haplotype restriction; and the simultaneous administration of MHC class I and class II epitopes which could prove beneficial for immunologic memory. In a meta-analysis, it was found that 8.1 % of patients vaccinated with whole-tumor antigen had notable clinical responses, while 3.6 % of patients vaccinated with molecularly defined tumor antigens had objective clinical responses (P < .001, χ 2 test) [106]. An intracavitary delivery of a viral oncolysate vaccine generated with ovarian cancer cell lines infected with influenza A virus showed objective clinical responses [107, 108]. Delivery of autologous tumor cells infected with Newcastle disease virus showed the same response [109]. HSV-infected tumor cells used directly or pulsed on dendritic cells have been shown to stimulate marked antitumor immune response in the mouse, which was better than utilizing ultraviolet-irradiated tumor cells [110–112]. Using mature dendritic cells (DCs) with whole autologous tumor lysate, 50 % of patients demonstrated remission inversion [113]. The use of DC/tumor cell fusion demonstrated to be able to induce antitumor cytotoxic T-lymphocyte activity in vitro [114]. There is a concern about the use of whole-tumor vaccination which relates to the inclusion of a large number of self-antigens, as it could drive tolerogenic responses (i.e., expand Treg) rather than cytotoxic lymphocyte responses. It has been shown that DCs can be polarized ex vivo with the use of interferons, Toll-like receptor agonists, or p38 mitogen-activated protein kinase inhibitors to stimulate cytotoxic lymphocytes and Th17. [115]. The limitation of cancer vaccines is due to the fact that it is unable to elicit an overwhelming T-cell response. In addition, it has been shown that immune modulation through blockade of the endothelin B receptor markedly increases the efficacy of weak vaccines by reversing the inhibitory function of tumor endothelium and enabling homing of tumor-reactive T cells [106, 116]. Moreover, it was observed that, depletion of Treg is a critical maneuver to enhance vaccine therapy [117].
Adoptive T-Cell Therapy
The advantages of TIL-based adoptive therapy include the presence of spontaneously occurring T cells with avidity against tumor which have escaped thymic deletion, the use of a polyclonal population of T cells, and the natural selection of patients whose tumor microenvironment is already conducive to T-cell homing. Incremental lymphodepletion through high-dose nonmyeloablating chemotherapy and added whole-body radiation was studied. Regression of metastatic tumors was observed, with 16 % complete response and 72 % objective response rates in recent reports with maximal lymphodepletion and radiation [118, 119]. In another study, it was shown that adoptive immunotherapy is a viable methodology and can control tumors of all sizes [120]. It was reported that patients who received adjuvant therapy with adoptive transfer of tumor-derived lymphocytes expanded ex vivo with IL-2 had a 3-year disease-free survival rate of 82.1 %, and the control group had 54.5 % [121, 122]. In melanoma trials, it was shown that TILs persisted 2 months after infusion in patients who exhibited tumor regression and were characterized by a less differentiated phenotype (CD27+ CD28+ CD45RA+ but CD62L− CCR7−) and longer telomeres [123–127]. It has been argued that the use of memory rather than effector cells may be more efficacious for adoptive transfer [128], and this was confirmed by mouse models [129]. June et al. [130] described the development of a next-generation K562-based aAPC platform capable of expressing multiple gene inserts, including human lymphocyte antigen (HLA) -A2, CD64 CD80, CD83, CD86, CD137L (4-1BBL), and CD252 (Ox40L), and several T-cell supporting cytokines. Rosenberg et al. [119] have demonstrated the clinical feasibility, safety, and preliminary efficacy of redirecting T cells of patients with melanoma using a TCR specific to the melanoma antigen MART-1. Adoptive transfer of TCR-transduced cells in 15 patients resulted in durable engraftment at levels exceeding 10 % of peripheral blood lymphocytes for at least 2 months after the infusion. In addition, high levels of circulating engineered cells at 1 year after infusion was observed in two patients who demonstrated regression of metastatic melanoma lesions. TCR-based engineering represents a reasonable strategy for ovarian cancer therapy as TCRs recognize HLA-A2 restricted epitopes from known ovarian cancer antigens such as NY-ESO-1 and p53 [131–134]. Optimization through selection of naturally occurring or recombinant high-affinity receptors, engineering to prevent recombination with endogenous TCR, and the use of lentiviral vectors have been developed [135]. Genetic modification to recognize antigens in an MHC-unrestricted fashion through the use of chimeric antigen receptors (CARs), fusion genes encoding an extracellular domain that specifically binds to tumor epitopes through a single-chain variable fragment (scFv) antibody, linked to intracellular signaling modules that mediate T-cell activation has been developed [130, 136, 137]. The tumor binding function of CARs is usually accomplished by the inclusion of a scFv antibody, containing the VH and VL chains joined by a peptide linker of about 15 residues in length. The universal targeting vectors can be constructed, as the scFvs bind to native cell surface epitopes and bypass the requirement for MHC restriction [138, 139]. Several CARs targeting diverse tumors have been developed [130, 140, 141, 142, 143], and some of the ovarian tumor antigens and CAR investigated in vitro and in vivo in T lymphocytes are FBP [144, 145], MUC-1 [145], HER2, and mesothelin [146].
Antibody-Based Immunomodulation
Modulating immune checkpoints by activation of effector cells, depletion of Tregs, or activation of professional APCs could improve the therapeutic efficacy of vaccines or adoptively transferred T cells. The development of functional antibodies has enabled effective immune modulation as shown in Fig. 12.2.
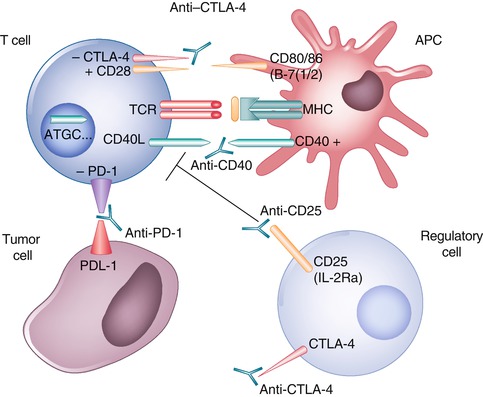
Fig. 12.2
Improved therapeutic efficacy of vaccines or adoptively transferred T cells can be achieved by modulating immune checkpoints, including activation of effector cells by blocking CTLA4 or PD-1; depletion of regulatory T cells by use of low-dose oral or intravenous cyclophosphamide or through targeting the interleukin-2 (IL-2) receptor α chain, also known as CD25; or activation of professional antigen-presenting cells (APCs) by stimulation with CD40 ligands
Dendritic Cell Activation
The main mechanism of immune stimulation by CD40 ligands is the activation of DCs resulting in increased survival, upregulation of molecules, and secretion of critical cytokines for T-cell priming. This promotes antigen presentation, priming, and cross-priming of CD4+ and CD8+ effector T cells. It was observed that agonistic anti-CD40 antibody is well used in combination with vaccines or Toll-like receptor agonists [147, 148], as it can accelerate the deletion of tumor-specific cytotoxic lymphocytes [149]. It was noted that ovarian cancers express the CD40 receptor [150–154] and respond to CD40 agonists with apoptosis and growth inhibition in vitro and in vivo [153, 155, 156].
Effector T-Cell Activation
T-cell activation is triggered through the T-cell receptor by recognition of the cognate antigen complexed with MHC. CTLA-4 blockade activates CD4 and CD8+ T effector cells by removing an inhibitory checkpoint on proliferation and function [157], and combination of direct enhancement of Teff cell function and inhibition of Treg activity is important for mediating the therapeutic effects of anti-CTLA-4 antibodies during cancer immunotherapy [158].
Most of clinical data to date have emerged from studies in patients with melanoma [159], where CTLA-4 blockade has yielded notable responses. It has been shown in patients with ovarian carcinoma, previously vaccinated with granulocyte–macrophage colony-stimulating factor modified, irradiated autologous tumor cells (GVAX) and received ipilimumab (1 month to 3 years after GVAX), that tumor regression correlated with the CD8+/Treg ratio [160]. PD-1 is a negative regulator of lymphocyte activation, which binds PD-L1 and PD-L2 ligands. PD-L1 is expressed on many tissues. Ovarian carcinoma cells, tumor-infiltrating tolerogenic DCs, and myeloid-derived suppressor cells express PD-L1 [161, 162]. It was observed that expression of PD-L1 by tumors conferred resistance to immunotherapy in mice [163], while antibodies blocking PD-L1 or PD-1 enhanced the efficacy of immune therapy [163, 164]. A phase I study using PD-1-blocking antibody showed the antibody to be safe and well tolerated in patients with hematologic malignancies [165].
Treg Depletion
CD4+ CD25+ Foxp3+ Treg is responsible for maintaining peripheral tolerance by inhibiting T-cell activity. A number of Treg-depleting strategies have been investigated. [119, 166–170]. One of these strategies was the use of low-dose oral or intravenous cyclophosphamide [118, 171]. It was shown that the use of anti-CD25 monoclonal antibody before vaccination led to complete tumor rejection and establishment of extended immunity with no side effects [118, 172]. Also, it was observed that administration of anti-CD25 resulted in transient reduction in CD4+ CD25+ Treg in patients with metastatic melanoma [173]. Denileukin diftitox, a fusion protein of IL-2 and diphtheria toxin that targets CD25-expressing cells was used in patients with melanoma, ovarian cancer, and renal cell carcinoma [174–177]. It was observed that these conjugates were immunogenic and induced neutralizing antibodies. In addition, daclizumab, which is humanized immunoglobulin G1-kappa monoclonal antibody that binds specifically to CD25 [178] has been used in autoimmune disorders [179, 180], acute graft-versus-host disease [181], and in patients with cancer with CD25+ T-cell malignancies [182]. The advantage of daclizumab is that it is well tolerated and has a half-life of 20 days [183]. Moreover, daclizumab was found to be compatible with effective vaccination [184]. As above, the assumption is that innovative immunotherapeutic strategies offer the promise of enhancing host antitumor responses which may improve clinical outcomes in women with ovarian cancer. While many preliminary phase I/II studies have demonstrated induction of antitumor responses, currently there is no clinically effective antigen-specific active immunotherapy available for patients with ovarian cancer. Ongoing investigation may help to define the role of immunotherapy alone or in combination with other treatment strategies in the treatment of ovarian cancer patients. These trials may determine a role for immunotherapy in the treatment of ovarian cancer.
Epithelial Growth Factor Receptor Targeting Therapy
The epithelial growth factor receptor (EGFR; also known as Her or ErbB) family of receptor tyrosine kinases has been shown to have oncogenic characteristics in several human cancers. It was suggested that members of the EGFR family, such as ErbB3, may have a role in promoting the growth and proliferation of human ovarian cancer cells. The EGFR family of receptor tyrosine kinases consists of four closely related family members: EGFR (Her1), ErbB2 (Her2), ErbB3 (Her3), and ErbB4 (Her4) [185]. These cytoplasmic membrane-bound receptors share a common extracellular ligand-binding domain and a single transmembrane domain, followed by an intracellular tyrosine kinase domain and a C-terminal non-catalytic signaling tail. Signaling through these receptors is typically mediated by homodimerization or heterodimerization with other family members. Binding of the ligand with the extracellular domain of its corresponding receptor induces a structural reconfiguration of the receptor, promoting exposure of dimerization domain [186, 187]. The EGFR family members can be activated by unphysiological stimuli (e.g., oxidative stress, UV, and γ-irradiation), by other receptor tyrosine kinases (MET, insulin-like growth factor-1 receptor, or tyrosine kinase receptor B), or by G protein-coupled receptors and adhesion proteins [188].
Epithelial Growth Factor Receptor (Her1)/ErbB2 (Her2) in Ovarian Cancer
Siwak et al., in a review [189], noted that epithelial growth factor receptor amplification and activating mutations have been reported in a small percentage of ovarian cancer cases (4–22 and <4 %, respectively). The EGFR overexpression rate varies from 9 to 62 %. Increased EGFR expression has been correlated with poorer patient outcomes. Several small molecule inhibitors that block EGFR kinase activity (e.g., gefitinib and erlotinib) have been explored. There are two phase II clinical trials on gefitinib (Iressa or ZD1839) as a single agent in treating platinum-refractory or -resistant ovarian cancer. No complete response was observed in either trial. In one trial, 37 % of the patients had stable disease for over 2 months while none of the patients had a partial or complete response. In the second trial, 4 out of 27 patients had progression-free disease for over 6 months, and one of these four patients had an objective response. Erlotinib (Tarceva, OSI Pharmaceuticals, Long Island, NY, USA), another small molecule inhibitor of EGFR, demonstrated a 6 % partial response rate in a multicenter phase II trial with EGFR-positive ovarian cancer patients with taxane- and/or platinum-refractory or -resistant disease. In a phase II study, 56 patients demonstrated no improvement in pathological CR rates compared with historical experience with erlotinib added to carboplatin and paclitaxel in the first-line therapy for ovarian cancer [189]. An additional study comparing the combination of erlotinib and the antiangiogenic agent bevacizumab also did not show any improvement compared with bevacizumab alone. Also, vandetanib, a small molecule inhibitor of VEGFR and EGFR signaling, did not demonstrate significant clinical benefit in recurrent ovarian cancer [190]. Monoclonal antibodies that bind directly to the extracellular domain of EGFR to block EGFR activation or reduce surface EGFR levels have also been tested. The objective clinical response of these therapeutic antibodies, such as cetuximab (Erbitux, ImClone LLC, Bridgewater, NJ, USA) and matuzumab (EMD7200), in ovarian cancer was limited [188]. Early studies suggested that Her2 overexpression in ovarian cancer was a frequent event; but, other studies using techniques validated in breast cancer suggest that Her2 overexpression and amplification frequency in ovarian cancer is a rarer event [191]. Overexpression of Her2 has been associated with poor prognosis in some studies but not others. This discrepancy may be explained by differences in the criteria and methods used to assess Her2 overexpression. A phase II trial with trastuzumab (Herceptin, Genentech, Inc., South San Francisco, CA, USA), a humanized anti-Her2 monoclonal antibody, reported a low (7 %) partial response rate with only a 2-month progression-free interval in recurrent ovarian cancers overexpressing Her2 [192]. In a phase II multicenter open-label trial, CI-1033 (Canertinib, Pfizer, Inc., New York City, NY, USA), an irreversible pan-EGFR family inhibitor, was shown to have no activity in unscreened patients who have failed previous platinum-based chemotherapy [193]. A phase I trial combining carboplatin and lapatinib (Tykerb, Tyverb, GlaxoSmithKline, plc, London, UK), an EGFR–Her2 dual inhibitor, was performed in unscreened patients with platinum-sensitive recurrent ovarian cancer [194]. In this trial, 27 % of patients had a partial response and another 27 % had stable disease.
Related posts:
 Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
Psychological Aspects of Hereditary and Non-hereditary Ovarian Cancer
 Management of Patients with Early-Stage Ovarian Cancer
Management of Patients with Early-Stage Ovarian Cancer
 Ovarian Cancer Screening and Early Detection
Ovarian Cancer Screening and Early Detection
 Treatment of Advanced Stage Ovarian Cancer
Treatment of Advanced Stage Ovarian Cancer
 Pathology of Non-epithelial Malignancies of the Ovary
Pathology of Non-epithelial Malignancies of the Ovary
 Surface Epithelial Tumours of the Ovary
Surface Epithelial Tumours of the Ovary
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


