Metastatic melanoma continues to place a substantial economic burden on western caucasian populations because of the disproportionately high incidence in young patients.1 Historically, the prognosis for patients with metastatic disease has been dismal with a median overall survival (OS) of 6 to 9 months and a 5-year survival rate of less than 10%.2 Until 2011, only dacarbazine and high-dose interleukin 2 (IL-2) were approved for the treatment of metastatic disease. Dacarbazine gained approval in 1975 for its modest response rate of 10% and median OS of 5.6 to 9.7 months.2,3 Attempts to improve on this with other cytotoxics (temozolomide or fotemustine), cisplatin-based combination chemotherapy, biochemotherapy, and concurrent use of targeted agents with dacarbazine resulted in marginally higher response rates, additional toxicity but no added survival advantage over single agent dacarbazine.3–6 High-dose IL-2 has consistently demonstrated an objective response rate (ORR) of 16% to 23% with durable complete responses seen in approximately 5% to 10% of patients.7 High-dose IL-2, however, requires skilled inpatient management, which has largely restricted clinical application to a small number of highly specialized centers.
Pleasingly, the outlook for patients with metastatic melanoma is rapidly improving with the advent of two distinct therapeutic approaches: (i) targeting key oncogenic driver mutations that underpin melanoma tumorigenesis and (ii) using immunomodulation to reverse immune suppression. Blocking the mitogen-activated protein kinase (MAPK) pathway with potent inhibitors of BRAF and MEK results in striking responses and a significant OS advantage;8–13 these combinations are now established as a new standard of treatment for patients with BRAFV600 mutant advanced melanoma. Ipilimumab, an anti-cytotoxic T-lymphocyte antigen-4 (CTLA-4) antibody results in durable responses in 20% of patients and an OS advantage for metastatic melanoma.14,15 Anti-programmed death 1 (PD1) and anti-programmed death ligand 1 (PDL-1) monoclonal antibodies are emerging as new treatments for melanoma with rapid, deep, and durable response in 40% of patients with an OS advantage.16–20 Many more novel agents and combinatory treatments intended to enhance efficacy and circumvent resistance are currently in preclinical and clinical development.
The past decade has witnessed major advances in understanding the biology and genomic landscape of melanoma. Melanoma is driven by initiating and driving oncogenic aberrations that cooperate with loss of tumor suppressor function to promote cancer proliferation and survival.21,22 Commonly, these include aberrations in oncogenes (BRAF, NRAS, CDK4, KIT, CCND1, ERBB4, AKT, NEDD9, RAC1, GNAQ, and GNA11), transcription factors (MITF, MYC, and ETV1), and tumor suppressors (CDKN2A, NF1, TP53, BAP1, and PTEN).23 These insights into underlying “driver mutations” coupled with therapeutic development have paved the way for highly effective genome-specific targeted therapies in this disease.
The RAS/RAF/MEK/ERK (MAPK) pathway (Fig. 18-1), implicated in regulating cell cycle, proliferation, and survival under normal conditions, is hijacked by oncogenic driver mutations (e.g., BRAF and NRAS mutations) or alterations in cell-surface receptors (e.g., KIT) in approximately 65% of melanomas, resulting in constitutive activation of MAPK signaling.24–28 Approximately 40% of cutaneous melanomas have mutations in BRAF27,29,30 and up to 80% of these BRAF mutations are characterized by the substitution of valine by glutamic acid at codon 600 (V600E) encoded on exon 15.27,31,32 Less frequent BRAF mutant genotypes includes V600K (substitution of valine by lysine) found in approximately 5% to 30%, V600R (substitution of valine by arginine) found in 1% to 7%, and V600D (substitution of valine by aspartic acid) found in 0.3%. Other BRAF mutations identified include K601E and L597.31–35 These mutations confer constitutive activation of BRAF similar to V600E. With routine molecular testing now integrated into standard of care, other rarer BRAF mutations are also increasingly identified; many of these have not been functionally characterized and may be oncogenic through RAF-dependent activation of MEK or alternatively through CRAF activation.31,33
FIGURE 18-1
The mitogen-activated protein kinase (MAPK) pathway and sites of frequent genomic alterations in melanoma. Under normal conditions, growth factors bind to cell surface receptor tyrosine kinases (RTKs) triggering downstream RAS-RAF-MEK-ERK (MAPK) and PI3K/AKT/mTOR signaling. This signaling controls cell proliferation, growth, and survival. Mutations in BRAF, NRAS, and KIT that are commonly found in melanoma result in constitutive activation of the MAPK pathway.
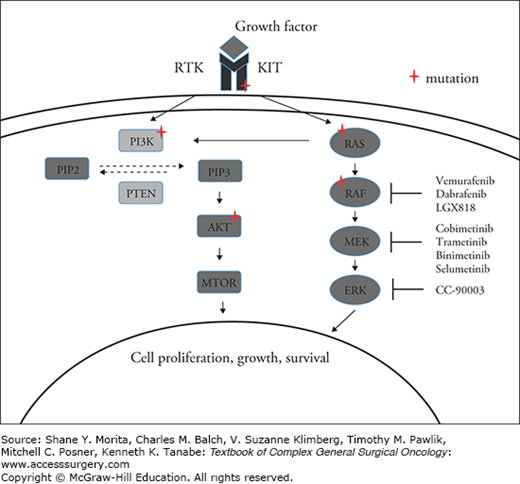
NRAS mutations, most commonly on codons 12 or 61, occur in 15% to 20% of melanomas and induce constitutive RAF signaling in the MAP kinase cascade.27,36,37 As BRAF is downstream of RAS, in the MAPK pathway, it is predictable that BRAF-V600E and NRAS mutations are mutually exclusive.38–40 NRAS mutations, however, can co-occur in non-V600 mutant melanoma.40 Although preclinical strategies to directly target KRAS do exist, equivalent approaches to inhibit NRAS are yet to be developed.41,42 Instead, therapeutic approaches aimed at targeting NRAS mutant melanoma have focused on combination strategies with PI3K and MEK inhibition (NCT01449058) or CDK4/6 and MEK inhibition (NCT01719380). A phase Ib study of the PI3Ka inhibitor, BYL719 combined with the MEK inhibitor binimetinib (MEK162) reported evidence of partial responses in a range of RAS mutant tumors including melanoma.43 Additionally, a phase 1b/2 study of the selective CDK4/6 inhibitor LEE011 in conjunction with binimetinib showed a highly promising ORR of 43% with a further 20% of patients achieving stable disease.44
KIT is a growth factor receptor tyrosine kinase that stimulates downstream MAPK and PI3K–AKT signaling. Activating mutations and/or amplification of KIT that confer kinase activity independent of ligand binding are commonly identified in acral (11% to 23%) and mucosal melanomas (15.6% to 21%).45,46 KIT mutant melanoma has been shown in several phase II studies to respond to the tyrosine kinase inhibitor imatinib with an overall objective response of 21% to 28%.46–49 Mutations in exon 11 and 13, especially L576P or K642E appear to be most predictive of response.46 Notably, a significant proportion of KIT mutant melanoma does not respond to imatinib implying that a subset of KIT mutations may not be the primary driver and that other mitogenic pathways may need to be targeted concurrently.46,47 Several phase II studies of second-generation tyrosine kinase inhibitors, such as nilotinib and dasatinib, have also demonstrated antitumor activity in KIT mutant melanoma.50,51
MEK is downstream of BRAF and is the major substrate of BRAF.52 This makes MEK a highly attractive target for therapeutic blockade in both BRAF and NRAS mutant melanoma. Recently, several phase II and III studies have provided clinical validation that combined BRAF and MEK inhibition enhances MAPK blockade with commensurate improvements in response rates and durability of response compared with single agent BRAF inhibitor.12,13,53
ERK is an additional node in the MAPK pathway. ERK inhibitors are currently entering phase I development (NCT02313012). It is possible that these agents will have an increasing role in both BRAF and NRAS melanoma. There is already preclinical evidence that ERK inhibitors have efficacy against BRAF wild-type and BRAF mutant melanoma cell lines, can overcome acquired BRAF inhibitor resistance, and show synergy with vemurafenib and MEK inhibitors in BRAF and NRAS mutant melanoma, respectively.54–56
The p16/cyclin D/CDK4/6/RB (CDK4) pathway that mediates cell cycle progression and inhibition of senescence and apoptosis is frequently dysregulated in melanoma.57–60 Loss of expression of the tumor suppressor CDKN2A (p16INK4A) via deletions, mutations, and transcription silencing is common in melanoma61–63 and frequently associates with mutations in BRAF.29,64 Less commonly, activation of the CDK4 pathway occurs via cyclin D1 (CCND1) amplification and cyclin-dependent kinase 4 (CDK4) mutation or amplification.29,65,66 CDK4 signaling cooperates with BRAF and NRAS mutations to promote tumor progression.67–70 Targeting CDK4 in conjunction with MAPK pathway inhibition is currently under evaluation.44
Deletions and mutations of the tumor suppressor, PTEN, results in downstream activation of PI3K/AKT signaling. PTEN loss co-occurs in 40% of BRAF mutation melanoma.29,38,64 In preclinical models, PTEN deletions cooperate with BRAF mutations to enhance tumorgenesis.22 Less commonly, v-akt murine thymoma viral oncogene homolog 3 (AKT3) amplification has been identified to overlap in BRAF mutant melanoma, further underscoring the coincident activation of BRAF and PI3K/AKT signaling in this disease. Rare occurrences of activating AKT3 mutations have also been reported.71 Concurrently targeting the MAPK and Pi3K pathway in melanoma-resistant models is effective,72 and this approach is currently being evaluated in early phase clinical development (NCT01512251).
Inactivation of p53 function via mutations or deletion or alternately by amplification of the p53 inhibitor MDM2 is uncommon in melanoma;73–76 however, increased expression of the p53 inhibitor MDM4 is observed in 65% of melanomas. In preclinical models, MDM4 expression enhances tumorigenecity when combined with mutations in NRAS and BRAF.77 MDM2 inhibitors that stabilize p53 are in phase I/II development as single agents and concurrently with targeting the MAPK pathway (NCT02110355).
There are striking distinctions in the prevalence and genotype of BRAF mutations based on patient age, anatomical location, and geographic regions.34 BRAF mutations are most prevalent in younger patients, in truncal primaries, and in superficial spreading and nodular histotypes. There is an inverse association between patient age and the prevalence of a BRAF mutation; the majority of patients under the age of 30 years have BRAF mutant melanoma while this is the case for only a quarter of those over the age of 70.32,34 Melanomas arising from the trunk and extremities, areas subjected to intermittent UV exposure are characterized by high rates of either BRAF or NRAS mutations (70%)29,34 and infrequently KIT mutation (2%).45,78 Although BRAFV600E mutation is most common, the prevalence of BRAF600K genotype increases with patient age and cumulative UV exposure such as primaries from the head and neck versus trunk or extremities and in regions with higher ambient UV exposure such as Australia, Florida, and Texas.34 Mucosal and acral melanoma that arises in anatomical locations protected from UV exposure exhibit low rates of BRAF mutation (3% to 14%) and higher rates of KIT mutation (10% to 39%).45,79,80 Uveal melanomas do not have BRAF, NRAS, or KIT mutations but are dominated by mutations in guanine nucleotide-binding protein Q (GNAQ) and GNA11 alpha-subunits of G-coupled protein receptors that also activate the MAP kinase and PI3K pathways.81 This clinical subcategorization based on key clinico-pathological features such as age, primary site, and histosubtype provides a useful conceptual framework for the initial patient consultation about potential therapeutic targets.
The pivotal discovery that over 50% of melanomas harbor activating mutations in the RAS/RAF/MEK/ERK pathway has led to focused efforts to target key nodes in this pathway (Fig. 18-1). The initial study with sorafenib, a nonselective type 2 RAF inhibitor was disappointing.82 Low response rates to sorefenib may, however, reflect an inability to achieve sufficient MAPK pathway blockade at tolerable doses and lack of patient preselection based on BRAF mutations. Clinical development of type 1 RAF inhibitors such as vemurafenib (PLX4032), dabrafenib (GSK2118436), and encorefenib (LGX818), all of which exhibit high selectivity for V600-mutant BRAF relative to wild-type BRAF has resulted in more complete and sustained inhibition of MAPK signaling at tolerable doses with striking clinical benefits observed in BRAF mutant melanoma.8,10
Vemurafenib was the first of these potent and selective BRAF inhibitors to enter clinical development (Table 18-1). Vemurafenib targets the kinase domain of mutant BRAF and results in diminished tumor growth through reductions of phosphorylated ERK.83 The landmark phase I dose-escalation trial of vemurafenib established the recommended phase 2 dose (RP2D) at 960 mg twice daily based on profound inhibition in phosphorylated ERK levels, corresponding inhibition of [18F] fluorodeoxyglucose uptake on positron emission tomography and promising efficacy as determined by tumor regression.84,85 The ORR was 81% and the median progression-free survival (PFS) was 7 months in the expansion cohort of BRAF V600E mutant melanoma patients.86 Phase II data confirmed these findings with an overall response of 53% (70 of 132 patients: 6% complete response and 47% partial response), a median PFS of 6.8 months, and a median OS of 15.9 months.87 The phase III registration study of vemurafenib versus dacarbazine (Table 18-1) undertaken in 675 treatment naive BRAFV600E/K mutant melanoma patients showed consistent results with an ORR of 59% versus 11% in favor of vemurafenib. Complete responses occurred in 5% of patients. The median PFS and OS were dramatically improved at 6.9 months versus 1.6 months (HR for PFS, 0.38; 95% CI, 0.32 to 0.46; P < 0.0001) and 13.6 months versus 9.7 months (HR for OS, 0.70; 95% CI, 0.57 to 0.87; P = 0·0008), respectively, for the vemurafenib group compared with the dacarbazine group.8,9 Comparable antitumor efficacy was observed in BRAFV600E and BRAFV600K mutants. Vemurafenib gained the Food and Drug Administration (FDA) approval for treatment of advanced V600E mutant melanoma on the basis of this phase III study.
Phase III Studies of BRAF and MEK Inhibitor Blockade in Melanoma
| Study Design | Number of Patients | Primary End Point | Treatment Arms | Response (ORR) | Median PFS | Median OS | HR (95% CI) for Primary End Point | Reference |
|---|---|---|---|---|---|---|---|---|
| Phase III, randomized | 675 | OS |
|
|
|
| (HR, 0.70; 95% CI, 0.57to 0.87; P = 0·0008) | BRIM3 [8,9] |
| Phase III, randomized | 250 | PFS |
|
|
| NA | (HR, 0.35; 95% CI, 0.20 to 0.61; P<0·0001) | BREAK3 [10] |
| Phase III, randomized, double-blind | 495 | PFS |
|
|
|
| (HR, 0.51; 95% CI, 0.39 to 0.68; P<0.001) | [13] |
| Phase III, randomized, double-blind | 423 | PFS |
|
|
|
| (HR, 0.75; 95% CI, 0.57 to 0.99; P = 0.03) | [12] |
| Phase III, Randomized, double-blind | 704 | OS |
|
|
|
| (HR, 0.69; 95% CI, 0.53 to 0.89; P = 0.005) | [53] |
Dabrafenib, a reversible, ATP-competitive type I BRAF inhibitor, is comparable to vemurafenib in both pharmacodynamic effect and antitumor potency. The phase I study established the RP2D at 150 mg twice daily and demonstrated an ORR of 50% (18 of 36 patients).88 In the phase II study, 92 patients with either BRAFV600E (72 patients) or BRAFV600K (16 patients) mutation positive metastatic melanoma were enrolled. The response rate, median PFS, and OS were 59% (45 of 76 patient), 6.3 months, and 13.1 months for the BRAFV600E group and 13% (2 of 16 patients), 4.5 months, and 12.9 months for the BRAFV600K group.89 In the phase III registration study (Table 18-1), 250 treatment naive BRAF V600E mutation-positive stage IV or unresectable stage III melanoma patients were assigned in a ratio 3:1 to either dabrafenib (187 patients) or dacarbazine (63 patients). Patients on the chemotherapy arm were permitted to cross over at the time of disease progression. A significantly higher ORR (50% compared with 7%) and longer median PFS (6.7 months compared with 2·9 months, HR, 0.35; 95% CI, 0.20 to 0.61; P<0.0001) were seen in the dabrafenib arm compared with the chemotherapy arm.10 Although direct comparisons have not been made, evidence suggests that vemurafenib and dabrafenib have comparable antitumor activity. Both drugs elicit a response rate in excess of 50%, a median PFS of approximately 7 months, and a median OS of approximately 13 months.9,10 Additionally, both agents have also demonstrated antitumor efficacy in patients with active brain metastasis.90,91
Overall vemurafenib and dabrafenib are well tolerated and toxicities are easy to manage. BRAF inhibitor class toxicities common to both agents include alopecia, keratoderma, keratotosis piliaris, papillomas, keratoacanthomas, and squamous cell carcinomas (SCC). The incidence of cutaneous SCC was 24% with vemurafenib and 5% with dabrafenib.8–10,92 These SCC occur as a consequence of formation of BRAF:CRAF heterodimers that cause paradoxical MAPK pathway activation in keratinocytes with upstream mutations in the MPAK pathway, most commonly activating mutations in HRAS.93,94 The oncogenic potential of BRAF inhibitor therapy in other neoplastic lesions with wild-type BRAF and activating RAS mutations has recently been highlighted in isolated cases of RAS mutant colorectal cancer and leukemia.95,96 These reports serve as a reminder for the need for long-term safety monitoring and clinical vigilance for secondary malignancies, especially in long-term responders or when these agents are used in the adjuvant setting. Toxicities more commonly seen with vemurafenib include arthralgia, photosensitivity rash, and elevated liver-enzyme levels, while pyrexia and chills are more common with dabrafenib.
Several MEK inhibitors such as trametinib (GSK1120212), binimetinib (MEK162), cobimetinib (GDC-0973), and selumetinib (AZD6244) have entered clinical development for melanoma. Among these, trametinib, an oral small-molecule, selective potent allosteric inhibitor of MEK1 and MEK2 is furthest along in development.97 The phase I study of trametinib revealed tumor responses and disease stabilization in V600E or V600K BRAF mutant melanoma.98 The phase III METRIC trial randomized 322 patients with metastatic BRAF V600E or BRAF V600K melanoma to receive trametinib or chemotherapy (either dacarbazine or paclitaxel) in a 2:1 ratio.99 Patients receiving chemotherapy were permitted to cross over to trametinib at documented disease progression. The confirmed response rate for trametinib was 22% (47 of 214 patients). The primary endpoint of median PFS was 4.8 months versus 1.5 months for the trametinib group compared with chemotherapy (HR for disease progression or death in the trametinib group, 0.45; 95% CI, 0.33 to 0.63; P<0.001). The 6-month OS rate was 81% in the trametinib group and 67% in the chemotherapy group (HR for death, 0.54, 95% CI, 0.32 to 0.92; P = 0.01), even though 51 of 108 patients (47%) receiving chemotherapy had crossed over to receive trametinib. Like the other MEK inhibitors, the toxicity profile for trametinib includes papulopustular rash, dermatitis acneiform, diarrhea, and infrequently ventricular dysfunction and reversible ocular events such as MEK-inhibitor–induced central serous retinopathy.
Despite rapid and impressive initial responses from BRAF inhibitors, durability in the majority is limited by the emergence of acquired drug resistance.9,10 Multiple mechanisms of resistance have been characterized. It is noteworthy that between 70% and 79% of melanomas progress via restoration of MAPK signaling.100–102 Although alterations to the drug-binding site of the BRAF protein do not occur, BRAF amplification103 and BRAF splice-variants capable of forming active BRAF dimers feature as prominent mechanisms of acquired MAPK reactivation.104,105 Other MAPK-dependent pathways identified include activating NRAS mutations,40,106 mutations in MEK,107,108 and overexpression of the MEK-activating kinase COT1, which activates ERK.109 Several MAPK-independent pathways of escape have also been described, the most dominant of which involves upregulating PI3K–PTEN–AKT–signaling via loss of PTEN, mutations in PI3K, PTEN, AKT1, and PHLPP1.100,102,106,108,110 Activation of receptor tyrosine kinases including platelet-derived growth factor receptor b (PDGFRb)106, insulin-like growth factor IR (IGF-IR)111, FGFR3112, and MET27,113 are also implicated in driving alternative signaling pathways (e.g., PI3K pathway) and mediating resistance.
Vemurafenib and dabrafenib significantly improved response rates, PFS and OS relative to chemotherapy in BRAF mutant melanoma.8,10 Nonetheless, the development of treatment resistance limits the durability of these responses to a median of 6 to 7 months.100–102,114 Additionally, BRAF inhibitor monotherapy has the liability of toxicity associated with paradoxical activation of the MAPK pathway.93,94 MEK inhibitors block MAPK signaling downstream of BRAF and thereby have the ability to enhance MAPK blockade and delay the onset of MAPK-dependent resistance when given in conjunction with a BRAF inhibitor. Sequential approaches of adding a MEK inhibitor in patients who had failed a prior BRAF inhibitor showed modest efficacy.115 Preclinical and clinical data supports upfront concurrent MEK and BRAF inhibition for enhanced MAPK pathway blockade, inhibition of tumor growth, delayed onset of MAPK-driven resistance, and diminished side effects that occur owing to paradoxical activation of the MAPK pathway in BRAF wild-type cells.11,93,115–117
The addition of trametinib, a MEK inhibitor, to dabrafenib was postulated to prevent or delay the onset of resistance. The initial phase I study showed an improved response rate from combined dabrafenib and trametinib therapy compared with dabrafenib monotherapy; however, the response rate in patients who have previously failed prior BRAF inhibitor therapy was less than 20%.118 In the phase II study, three dosing schedules of dabrafenib and trametinib (75 mg/1 mg, 150 mg/1.5 mg, and 150 mg/2 mg) were evaluated in 247 patients with BRAFV600 mutation metastatic melanoma.11 This study established that dabrafenib and trametinib at full dose (150 mg/2 mg) achieved the best efficacy without any additional toxicity. The dabrafenib 150 mg twice daily and trametinib 2 mg daily cohort resulted in an improved median PFS of 9.4 months as compared with 5.8 months in the monotherapy group (HR, 0.39; 95% CI, 0.25 to 0.62; P<0.001) leading to FDA approval. The ORR and complete response rate for the 150 mg/2 mg combination group as compared with dabrafenib monotherapy were 76% versus 54% (P = 0.03) and 9% versus 3%, respectively. In a phase III study of dabrafenib and trametinib versus trametinib, 423 untreated patients were enrolled (Table 18-1). The median PFS was 9.3 months for dabrafenib as compared with 8.8 months for trametinib (HR, 0.75; 95% CI, 0.57 to 0.99; P = 0.03). The ORR was 67% for the combined group versus 51% for the dabrafenib only group (P = 0.002). At 6 months, the interim OS rate was 93% for dabrafenib–trametinib and 85% with dabrafenib alone (HR, 0.63; 95% CI, 0.42 to 0.94; P = 0.02).12 A subsequent phase III trial compared dabrafenib and trametinib versus vemurafenib in 704 patients with BRAF V600E or V600K mutation positive metastatic melanoma as first-line therapy (Table 18-1). OS was the primary endpoint. The 12-month OS was 72% in the combination-therapy group and 65% in the vemurafenib group (HR for death, 0.69; 95% CI, 0.53 to 0.89; P = 0.005).53 The median OS was 17.2 months for patients in the vemurafenib group and had not been reached for patients in the combination-therapy group. An advantage in favor of the dabrafenib and trametinib combination compared with vemurafenib was also noted in terms of median PFS (11.4 months vs. 7.3 months: HR, 0.56; 95% CI, 0.46 to 0.69; P<0.001), objective response (64% vs. 51%, P<0.001), median duration of response (13.8 months vs. 7.5 months), and complete response rate (13% vs. 8%). Specifically in the BRAF V600K subgroup, the response rates for dabrafenib and trametinib compared to vemurafenib was 65% and 44%, respectively. Common adverse events from the combination included pyrexia, nausea, diarrhea, chills, fatigue, headache, and vomiting. Skin side effects such as rash (43% vs. 22%), photosensitivity reaction (22% vs. 4%), hand–foot syndrome (25% vs. 4%), skin papillomas (23% vs. 2%), SCC and keratoacanthomas (18% vs. 1%), and hyperkeratosis (25% vs. 4%) were more common in the vemurafenib group compared with the dabrafenib and trametinib combination. In contrast, pyrexia was more frequent in the combination-therapy group than in the vemurafenib group (53% vs. 21%).
The combination of the BRAF inhibitor, vemurafenib and MEK inhibitor, cobimetinib was first tested in a phase Ib trial that enrolled 66 vemurafenib-refractory patients and 63 BRAF-inhibitor-naive patients with BRAFV600 mutation-positive metastatic melanoma. The maximum tolerated dose (MTD) was established at vemurafenib 960 mg, twice a day in combination with cobimetinib 60 mg daily, 21 days on and 7 days off. Efficacy endpoints were significantly enhanced by the addition of cobimetinib. In BRAF inhibitor naive patients, the objective response was 87% (55 out of 63) with a median PFS of 13.7 months. The complete response rate was 10% (6 out of 63), which was double than previously observed for vemurafenib monotherapy. In the vemurafenib refractory cohort, responses were less impressive with a response rate of 15% (10 out of 66) and a median duration of response of 6.7 months.119 The phase III registration study of vemurafenib and cobimetinib versus vemurafenib and placebo in 495 previously untreated patients has set a new treatment benchmark for patients with BRAFV600E mutation-positive advanced melanoma.13 This landmark study reported a median PFS of 9.9 months for the combination group compared with 6.2 months for single agent vemurafenib (HR, 0.51; 95% CI, 0.39 to 0.68; P<0.001). Objective response and complete response rates were 68% versus 45% (P<0.001) and 10% versus 4% for the combination group compared with single agent vemurafenib. The 9-month OS rate was promising at 81% in the combination group versus 73% in the control arm. Overall, vemurafenib and cobimetinib were not associated with a significant increase in grade 3 adverse events over single agent vemurafenib. Common adverse events for the combination were non-acneiform rash, diarrhea, fatigue, photosensitivity, liver enzyme abnormalities, and reversible MEK-inhibitor–induced serous chorioretinopathy. Notably, the incidence of cutaneous SCC associated with paradoxical MAPK signaling was reduced from 11% to 2% with the the addition of the combination of BRAF and MEK inhibition.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


