Synovial cell sarcoma represents a rare group of cancers, particularly in the head and neck region, that typically affects young individuals and has a male preponderance. Prognosis varies with patient age, site and size of the malignancy, degree of necrosis, high level of mitotic activity, and neurovascular invasion. Complete surgical resection of the tumor via partial or total laryngectomy is the first-line treatment in locally invasive disease. CO 2 lasers have been shown to be useful in controlling localized disease. There is also a role for adjuvant radiotherapy. Ifosfamide-based chemotherapy is most useful for malignant disease.
Key Points
- •
Synovial sarcoma of the larynx represents a rare entity, with challenging diagnosis and management issues.
- •
A multidisciplinary therapeutic approach is essential for management of this malignancy.
- •
Long-term follow-up is essential to monitor for recurrence and improve disease-free survival.
Introduction
Synovial cell sarcomas are rare, aggressive, mesenchymal malignancies constituting a high-grade histologic variety of sarcomas. These tumors represent the fourth most common type of sarcoma after malignant fibrous histiocytomas, liposarcomas, and rhabdomyosarcomas. Synovial cell sarcomas predominantly affect soft tissues of the extremities such as large joints, bursal structures, and tendon sheaths, particularly in adolescents, among whom they account for 10% of all soft-tissue sarcomas. This malignancy has preponderance in males with a male/female ratio of 1:2. Approximately 3% of synovial cell sarcomas can also occur in the head and neck region, which is poor in synovioblastic tissue, and fewer than 100 cases in this region have been described in the literature to date. It has been reported to occur in various sites within the head and neck, most commonly the hypopharynx. Other sites include the oropharynx and soft tissues of the neck. However, synovial cell sarcomas of the larynx are extremely rare, with only a handful of cases reported to date. As a result it is not often diagnosed until later in the course of the disease, which therefore affects management and prognosis. Being such a rare entity, the treatment of head and neck synovial cell sarcomas is not well researched and classified.
Introduction
Synovial cell sarcomas are rare, aggressive, mesenchymal malignancies constituting a high-grade histologic variety of sarcomas. These tumors represent the fourth most common type of sarcoma after malignant fibrous histiocytomas, liposarcomas, and rhabdomyosarcomas. Synovial cell sarcomas predominantly affect soft tissues of the extremities such as large joints, bursal structures, and tendon sheaths, particularly in adolescents, among whom they account for 10% of all soft-tissue sarcomas. This malignancy has preponderance in males with a male/female ratio of 1:2. Approximately 3% of synovial cell sarcomas can also occur in the head and neck region, which is poor in synovioblastic tissue, and fewer than 100 cases in this region have been described in the literature to date. It has been reported to occur in various sites within the head and neck, most commonly the hypopharynx. Other sites include the oropharynx and soft tissues of the neck. However, synovial cell sarcomas of the larynx are extremely rare, with only a handful of cases reported to date. As a result it is not often diagnosed until later in the course of the disease, which therefore affects management and prognosis. Being such a rare entity, the treatment of head and neck synovial cell sarcomas is not well researched and classified.
Epidemiology
Synovial cell sarcoma is a rare malignancy with an incidence of 2.75 per 100,000. The incidence in the head and neck is significantly lower, with approximately 80 cases of head and neck synovial sarcomas reported to date. Laryngeal synovial cell sarcomas are extremely rare, with about 20 cases reported in the literature.
Synovial cell sarcoma is primarily a malignancy affecting adults in the third to fifth decade of life. In the head and neck region, synovial cell sarcoma is noted to occur during this age range as well, although it can also occur outside of this range. Of the specific cases of laryngeal synovial cell sarcoma, the youngest known patient was 14 and the oldest 79 years old. Furthermore, males are predisposed to this malignancy more than females, with a male/female ratio of 2:1.
Etiology
Synovial sarcomas are thought to arise from pluripotent mesenchymal cells that have the ability to differentiate into epithelial and mesenchymal lineages. Despite the name, synovial cell sarcomas can arise from cells outside of the synovium as exemplified by synovial cell sarcomas arising in the parapharyngeal, hypopharyngeal, or laryngeal regions.
The underlying defect in synovial cell sarcoma is primarily a specific chromosomal translocation t(X;18)(p11;q11). This translocation creates a so-called fusion gene SYT-SSX1 , SYT-SSX2 , or SYT-SSX4 . The SYT component arises from chromosome 18 and the SSX gene arises from chromosome X.
Pathogenesis
The pathogenesis of this tumor involves the creation of aberrant transcriptional regulators resulting in the inhibition of tumor suppressor genes or the activation of proto-oncogenes. The SYT component (chromosome 18) fuses with homologous genes at Xp11 with 1 of 3 sites: SSX1, SSX2, or SSX4. Monophasic tumors may have any of the transcripts, but biphasic tumors predominantly express the SYT-SSX1 transcript.
Pathologic findings
Macroscopic Pathology
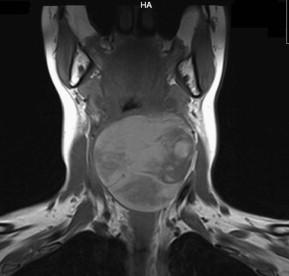
On gross appearance, no distinguishing characteristics have been described for synovial cell sarcomas. The tumors may have a gray-white almost ivory appearance with an oleaginous texture on palpation. In the head and neck, synovial cell sarcomas most commonly occur in the hypopharynx and very rarely in the larynx. Laryngeal synovial cell sarcomas have been known to occur in the supraglottis, glottis, and subglottis. Macroscopically, synovial cell sarcomas may have a cystic, solid, or necrotic appearance. In the upper airways, these cancers may appear as exophytic lesions with superficial ulceration. Fig. 1 depicts the gross appearance of a laryngeal synovial cell sarcoma encountered by the authors.
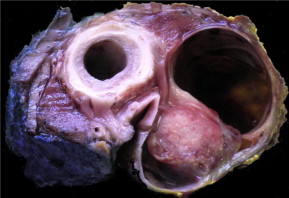
Histopathology
Up to 3 subtypes of synovial cell sarcomas have been described in the literature: monophasic, biphasic, and poorly differentiated.
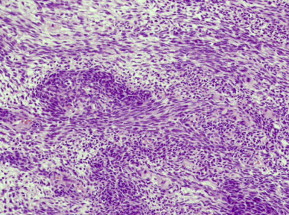
The monophasic and biphasic subtypes are the 2 main histologic subtypes of synovial cell sarcomas. Histologically both subtypes have proliferating spindle cells and branching, dilated, thin-walled blood vessels (hemangiopericytomatous pattern) within a heterogeneous collagenous stroma. Tumor necrosis, cystic changes, and calcification are other features recognized in synovial cell sarcomas.
The monophasic subtypes usually contain only spindle cells with a high nuclear to cytoplasm ratio, and tend to grow in a fascicular pattern. Alternatively, although this subtype does not have the tendency to develop epithelial-like cells, it may occasionally develop with solely an epithelioid component. Owing to its appearance, this subtype poses a diagnostic problem and can be misdiagnosed as a spindle cell sarcoma or a hemangiopericytoma.
The biphasic subtype varies, and contains glandular structures in addition to all the other histologic features. Furthermore, it contains clustering epithelioid structures intermixed with spindle cells, giving it a biphasic appearance microscopically. Figs. 2 A, B and 3 depict a biphasic laryngeal synovial cell sarcoma. Gland formation can be identified using reticulin staining and immunohistochemistry.
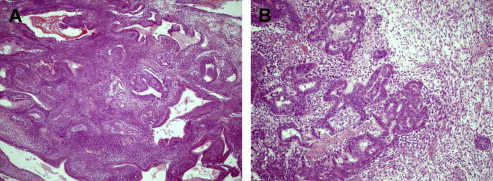
The third and more recently identified form is the poorly differentiated synovial sarcoma. This subtype is composed of uniform, densely packed, small cells. Poorly differentiated synovial sarcoma histology may be interspersed with other histologic subtypes, therefore posing a diagnostic dilemma.
Molecular Pathology
Synovial cell sarcomas contain the (X; 18) (p11.2; q11.2) translocation, which is specific to this malignancy and is present in 99% of the cases. This translocation results in the fusion of SYT genes located on autosomal chromosome 18 with highly homologous SSX-1, SSX-2, and SSX-4 genes on the X chromosome. Identification of this translocation plays an important role in diagnosing synovial cell sarcoma. The SYT-SSX-1 fusion gene occurs more commonly than the SYT-SSX-2 fusion gene, in a ratio of 3:2. The SYT-SSX4 combination is rare. The SYT-SSX1 fusion gene is mainly found in the biphasic synovial cell sarcoma subtype, whereas monophasic and poorly differentiated forms contain a mixture of SYT-SSX1 and SYT-SSX-2 translocations. Based on this observation, Eilber and Dry suggest that SSX1 proteins promote epithelial cell differentiation whereas SSX2 proteins inhibit the same.
Clinical presentation
Synovial cell sarcomas tend to present with nonspecific symptoms and vary with location of the tumor. Several studies have reported asymptomatic presentation of these cancers. However, among symptomatic patients the most frequently reported symptoms include dysphagia, dyspnea, dysphonia, pain, and facial mass.
Diagnostic workup
Diagnosis of a suspected laryngeal synovial cell sarcoma involves obtaining a thorough history of presentation, clinical examination, and meticulous tumor staging. These investigations include radiologic imaging, biopsy of the lesion, immunohistochemistry to obtain a histologic diagnosis, and molecular testing.
Radiologic investigation of the suspected malignancy includes computed tomography (CT) and magnetic resonance imaging (MRI) to identify the exact size, location, and extent of the malignancy, as well as invasion of adjacent structures ( Figs. 4 and 5 ).
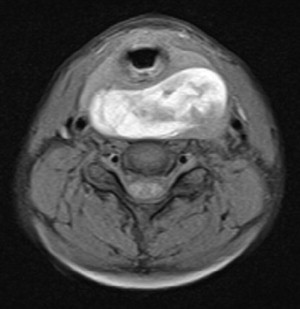
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


