Fig. 30.1
Margins used in radiation treatment. GTV gross tumor volume, CTV clinical target volume, PTV planning target volume
One rapidly evolving area is in the treatment of patients with metastatic disease. Radiation is commonly delivered to palliate symptoms such as pain, bleeding, or neurological symptoms from mechanical compression of growing tumors. In this setting, shorter regimens have shown similar efficacy with the endpoint of symptom control. For example, the landmark palliative study RTOG 97-14 randomized breast and prostate cancer patients with painful bony metastases to 30 Gy in 10 fractions compared to 8 Gy given in a single fraction and showed equivalent pain relief [5]. This has led the American Society for Radiation Oncology (ASTRO) to recommend this single-fraction treatment in appropriately selected patients for pain relief [6].
In addition to shortening treatment schemes, reduction or elimination of CTV margins is implemented when treating metastases. Improved immobilization and imaging techniques assure adequate coverage of the target with a precision that matches that of surgery and enables much less normal tissue in the field. Safe delivery of doses in the range of 6–30 Gy or higher, over 1–5 fractions, can be achieved, defined as stereotactic radiation treatments (stereotactic radiosurgery or SRS, stereotactic body radiotherapy or SBRT, stereotactic ablative radiotherapy or SABR). For each dose erogation, the patient is immobilized and imaged in the treatment position to ensure the target is treated accurately. Stereotactic treatments are a promising way to deliver a higher dose per fraction with low toxicity rates compared to traditional fields: they are commonly used to treat metastases to the adrenal gland, spine, liver, lung, and brain in appropriately selected patients. One of the most standard applications of this newer technique is treating metastases to the brain, where spearing normal tissue is of particular importance. Prior to stereotactic treatments, patients with brain metastases were treated with radiation to the whole brain, generally with 30 Gy over 10 fractions. Whole brain radiation is associated with significant acute and chronic toxicity, including nausea, vomiting, hair loss, confusion, and cognitive decline. Stereotactic treatments that treat brain metastases result in minimal cognitive decline by sparing the normal brain. They are becoming the standard of care for an increasing number of patients, depending on the number and type of metastatic lesions and life expectancy. Supporting this trend, a recent individual patient data meta-analysis showed that for patients less than 50 years old, stereotactic treatment alone (without the addition of whole brain radiotherapy) was associated with improved survival [7].
The use of a single-fraction dose for stereotactic treatments to brain metastases was studied in RTOG 90-05, a trial of single-fraction dose escalation based on the size of brain metastasis [8]. While higher doses had minimal toxicity for small lesions (less than 2 cm), treatment of larger lesions was associated with neurotoxicity including radionecrosis (necrosis of brain tissue as a result of radiation). Thus, caution needs to be employed when delivering high doses in a single fraction, and many centers split the single dose into 3–5 fractions to reduce toxicity particularly for larger tumors.
30.3 From Local to Abscopal: Radiotherapy and Systemic Tumor Control
While the cytotoxic effects of radiotherapy are exquisitely local, occasional distant tumor regression outside of the irradiated field has been observed and defined by Mole in 1953 as abscopal effect (ab scopus, outside the target) [9]. Abscopal effects have been reported in a number of different malignancies but are rarely seen with radiation alone [10]. Interestingly, abscopal responses have been seen with increased frequency when radiotherapy was used in melanoma patients progressing during treatment with immune checkpoint inhibitors [11–14]. These clinical observations, together with accumulated experimental evidence that the abscopal effect is mediated by T cells [15, 16], have raised a lot of interest in the possibility of using radiation to increase responses to immunotherapy. As detailed in the following sections, many clinical trials are exploring combinations of radiotherapy with various immunomodulators.
It remains to be established if the ability of radiation to exert an abscopal effect is dependent on the intrinsic immunogenicity of a tumor. Dramatic abscopal effects have been observed in combination with ipilimumab in melanoma and non-small cell lung cancer (NSCLC) [13, 17], two malignancies with a high mutational burden and a higher likelihood of expressing neoantigens recognized by T cells [18, 19]. On the other hand, abscopal responses elicited by radiation alone have also been reported in patients with renal cell carcinoma, a tumor type with low mutational burden but a propensity to respond to immunotherapy [20, 21]. Moreover, abscopal responses elicited by radiation alone or in combination with different immunomodulators have been seen in a variety of other malignancies [22–30]. Overall, these data suggest that abscopal effects can be elicited by radiation across several tumor types. However, it is presently unknown if some degree of pre-existing antitumor immunity is required.
It has also been suggested that the location of the irradiated metastatic site determines the likelihood of achieving abscopal effects [31]. While it is conceivable that, due to tumor heterogeneity and organ-specific features, different metastases generate a more or less immunosuppressive microenvironment, there is currently no evidence to guide the choice of site to be irradiated to achieve abscopal responses [32].
Overall, extensive work in preclinical tumor models has demonstrated that radiation can elicit antitumor T cells that contribute to control of the irradiated tumor [33–35]. However, abscopal effects have been more difficult to achieve, probably because they require a broader and more robust antitumor T cell response, capable of recognizing multiple antigens to account for possible tumor heterogeneity and overcoming the barriers present within the microenvironment of nonirradiated metastases. For a given tumor, it is likely that the strength of the immune response induced by radiotherapy is determined by the immunotherapy agent and radiation regimen used. Experimental evidence in mouse models of breast and colorectal carcinoma unresponsive to immune checkpoint inhibitors showed abscopal responses only when radiation was added and suggested that the radiation regimen is critical: with antibody targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), a single dose of 20 Gy was ineffective, while 6 Gy X 5 and 8 Gy X 3, given in consecutive days, achieved complete and partial abscopal responses [16]. While in the clinic the best radiation regimen to be used in combination with immunotherapy remains undefined, it is intriguing that, at least in combination with anti-CTLA-4, abscopal responses were achieved with the use of radiation regimens similar to the ones effective in mice [13, 17, 36].
30.4 Augmenting Local Control: Radiotherapy Effects that Promote the Effector Phase of the Antitumor Immune Response
The observation that in mice lacking a normal T cell compartment, higher radiation doses were required to achieve cure of irradiated tumors when compared to immunocompetent mice first implicated T cells in the local response to radiation [33]. This early finding was confirmed experimentally three decades later with the use of mouse tumors bearing model antigens [34, 35]. It remains to be determined to which degree T cells contribute to local radiotherapy responses in patients. Model antigens like ovalbumin are convenient for proof-of-principle studies, but their high immunogenicity and levels of expression fail to mimic the reality of cancer patients, with tumors that have gradually evolved to escape the immune system.
In addition, radiotherapy also evokes immunosuppressive signaling. For instance, when two poorly immunogenic mouse carcinomas were treated with radiotherapy, CD8+ T cell responses to three endogenous antigens were seen only upon neutralization of TGFβ, a strongly immunosuppressive cytokine that is activated by radiation [37]. In patients, evidence of priming of tumor-specific T cells following standard-of-care radiotherapy alone is scant [38]. Thus, it is possible that the priming of T cell by radiation occurs only when radiation is combined with immunotherapy. Noticeably, in patients with a pre-existing antitumor T cell response [39], radiation can reactivate a stalled immune response by countering two key immune escape mechanisms at the effector phase of antitumor immunity: T cell exclusion from the tumor microenvironment and reduced cancer cells antigenicity due to downregulation of major histocompatibility complex class I (MHC-I) antigen-presenting molecules on cancer cells [40, 41] (Fig. 30.2).
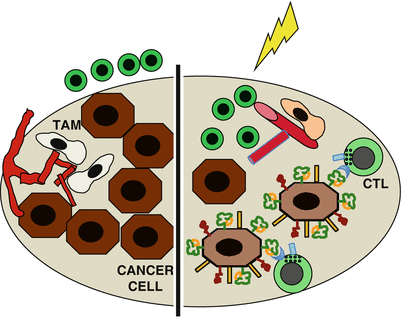
Fig. 30.2
Radiotherapy effects that promote the effector phase of the antitumor immune response. Left, cytotoxic T cells (CTL) are present but unable to enter the tumor due to the vascular barrier, which is promoted by pro-angiogenic tumor-associated macrophages (TAM). In addition, downregulation of MHC-I on the cancer cells precludes their recognition by CTL. Right, radiation reprograms the macrophages leading to vascular normalization, which allows tumor infiltration by CTLs. In addition, radiation upregulates MHC-I, NKD2D ligands, and Fas on the cancer cells, making them good targets for recognition and elimination by CTLs
Radiation-induced changes that counter T cell exclusion include release of chemokines that attract effector T cell to the tumor [42, 43] and increased expression of adhesion molecules on the vascular endothelium that facilitate T cell infiltration, as demonstrated in mouse breast and melanoma models [34]. In addition, reprogramming of tumor-associated macrophages (TAM) by radiation has been shown to cooperate with adoptive T cell transfer to allow rejection of pancreatic tumors by promoting vascular normalization [44]. Interestingly, increased infiltration of irradiated tumors by T cells activated by peripheral vaccination was also seen in an orthotopic mouse model of brain glioma, suggesting that it can occur in a variety of solid tumor types growing in different anatomical locations [45].
Upregulation of MHC-I expression by radiation has been shown in multiple mouse and human cancer cells in vitro and in vivo [45–48]. In vitro, increased MHC-I improved lysis of the irradiated cancer cells by CD8+ cytotoxic T cells (CTL), and in vivo it improved tumor rejection mediated by adoptively transferred and vaccine-activated T cells [45–47]. In addition to increasing the antigenicity of cancer cells, radiation has also been shown to increase the expression of stress-induced ligands that bind to the NK group 2, member D (NKG2D) receptor expressed by CTL and natural killer (NK) cells and promote killing of the cancer cells by these effectors [49–52]. Another way that radiation increases killing of cancer cells by T cells is by inducing the expression of death receptor Fas/CD95 [53].
Thus, multiple effects of radiation on the cancer cells and tumor microenvironment cooperate to enhance immune-mediated tumor rejection. Adaptive immunity may contribute to local tumor control achieved by radiotherapy if patients have pre-existing antitumor T cells [39]. In addition, tumor-targeted radiotherapy can sensitize resistant metastases to adoptively transferred T cells, as shown in preclinical studies [47, 53]. Radiation may also recover tumor responses to other immunotherapies that activate endogenous antitumor T cells. Downregulation of MHC-I in tumors is one of the mechanisms of resistance to multiple immunotherapies [54] including programmed cell death protein-1 (PD-1) blockade [55], and recent work in a mouse model of PD-1 resistant lung cancer suggests that at least in some cases, radiation-induced upregulation of MHC-I molecules on the cancer cells overcame resistance to anti-PD-1 treatment [56].
30.5 Inducing Abscopal Effects: Radiotherapy Effects that Promote Priming of Antitumor T Cells
Generating antitumor T cells in patients lacking such natural responses remains one of the major challenges in cancer immunotherapy. While presumably all cancers are recognized by the immune system during the course of their development, a majority of tumors that become clinically detectable escape by editing out the antigens recognized by T cells [57]. However, the genetic instability intrinsic to neoplasia fuels the generation of more mutations that can be antigenic and be targets of strong antitumor responses [58]. Rather a major barrier to development of antitumor T cells is exclusion and dysfunction of dendritic cells [59–61].
Radiation has the ability to overcome, at least in part, this major barrier to the priming of antitumor T cells (Fig. 30.3). There is evidence in experimental models that radiation increases the recruitment of DCs to the tumor and their activation, mediated by danger signals that are released during immunogenic cell death (ICD) induced by radiation [62–65]. Well-characterized danger signals released during ICD include calreticulin translocation to the surface of the dying cells that promotes their uptake by DCs, release of high-mobility group box-1 (HMGB-1) that binds to Toll-like receptor 4 (TLR4) on DCs, and ATP that activates the inflammasome in the DCs downstream to P2XR7 receptor [63, 66, 67]. In addition, when DNA from the irradiated cancer cells finds its way to the cytosol of DCs, it activates the production of interferon type I (IFN-I) via the stimulator of interferon genes (STING) pathway providing another signal that acts in autocrine fashion to activate DCs [68].
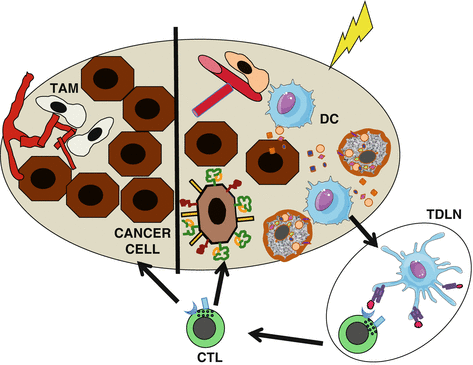
Fig. 30.3
Radiotherapy effects that promote priming of antitumor T cells. Right, danger signals generated by irradiated cancer cells drive dendritic cell (DC) recruitment to the tumor, where they uptake dying cancer cells and become activated. Activated DCs carry the antigens to the tumor-draining lymph node (TDLN) where they cross-prime tumor-specific T cells. Primed T cells become cytotoxic T cells (CTL) that go to both the irradiated tumor (right) and nonirradiated metastases (left) mediating the complete regression of the irradiated tumor and abscopal responses
Generation of the above signals depends on stress response pathways activated, while cancer cells are dying, including the unfolded protein response and autophagy [69]. Cell death after irradiation of cancer cells occurs in a variety of ways, depending on which survival/apoptosis pathways are activated, as well as the type of tumor microenvironment treated and the radiation dose and fractionation used [70]. Importantly, the availability of DCs limits the magnitude of antitumor T cell responses primed by radiation, and the activation of immunosuppressive mechanisms hinders the process [37, 71]. Thus, while radiation can convert the tumor into an immunogenic hub, it tends to provide a suboptimal vaccination. In the clinic most established cancers are poorly immunogenic. To successfully achieve priming of robust antitumor T cell responses, they require immunotherapies that block suppressive pathways or enhance immune stimulation [72]. Successful combinations have been achieved in preclinical studies with immune checkpoint inhibitors targeting CTLA-4, PD-1, and PDL-1, as well as agonistic antibodies to costimulatory receptor CD137 and TLR agonists [73–79].
The issue of whether radiotherapy induces neoantigens remains unsettled. In most studies, priming of T cell responses by radiation used alone or in combination with immunotherapy has been measured for a single antigenic epitope, often an immunodominant endogenous antigen or an exogenous reporter antigen introduced into the cancer cells (e.g., ovalbumin, OVA). To obtain a broader view of CD8+ T cell responses elicited by radiation, we have analyzed responses to four distinct epitopes derived from three antigens that are overexpressed in cancer cells, the anti-apoptotic protein survivin, the epithelial-to-mesenchymal transition (EMT) transcription factor Twist, and the envelope of an endogenous retrovirus gp70. Responses to all four epitopes were coordinately elicited by radiation of the poorly immunogenic mouse 4T1 mammary carcinoma when inhibition of radiation-induced DC activation was prevented by TGFβ neutralization [37]. Thus, removal of critical immunosuppressive blocks unleashes the potential of radiation to elicit a broad T cell response. Work is ongoing to comprehensively evaluate the repertoire of T cell clones primed following radiation.
In addition to antigenic specificity, the number and persistence of the antitumor T cells primed by radiation are likely to be critical for abscopal responses. In fact, abscopal responses are rarely seen despite the fact that the irradiated tumor is successfully rejected and represent a more stringent test of effective in situ vaccination by radiation [80]. As mentioned above in Sect. 30.3, the dose of radiation and its fractionation seem to be critical determinants of the ability of radiation to induce abscopal responses in combination with anti-CTLA-4. Doses of 6–8 Gy repeated three to five times achieved abscopal responses but not a single higher radiation dose of 20 Gy [16]. Recent data indicate that the difference between effective and ineffective radiation regimens is due to their differential ability to induce cancer cell-intrinsic activation of IFN-I pathway (Vanpouille-Box, Formenti, and Demaria, manuscript submitted). Overall, the optimal radiation regimens when radiotherapy is harnessed as an adjuvant for immunotherapy may be distinct from those employed in the practice of radiation oncology, an issue of paramount importance for clinical translation [4].
30.6 A Balancing Act: Negative Regulators of Antitumor Immunity Elicited by Radiation in the Tumor Microenvironment
Tumor escape from immune-mediated control that allows tumor progression involves multiple immunosuppressive pathways which are variably utilized by each given tumor [81]. While radiation, as discussed above, can mitigate or counter some of them, it also exacerbates others [82]. Acute activation of latent TGFβ by radiation is due to its dissociation from the latency-associated peptide mediated by a ROS-induced conformational change of the latter [83, 84]. TGFβ is a strongly immunosuppressive cytokine with effects on multiple immune cells [85]. For instance, activation of naïve CD4+ T cells in the presence of TGFβ facilitates their conversion into regulatory T cells [86, 87]. It remains unclear if radiation-induced TGFβ activation contributes to the radiation-induced increase in regulatory T cells that has been reported in some studies [88, 89]. On the other hand, as discussed above, inhibition of DC activation by TGFβ is a clear barrier to radiation-induced priming of antitumor T cells [37].
Increased recruitment of myeloid cells to the tumor has been shown to be driven by radiation-induced upregulation of colony-stimulating factor 1 (CSF1) in prostate cancer [90] and by C-C motif ligand 2 (CCL2) in pancreatic adenocarcinoma [91]. The recruited myeloid cells differentiated into immunosuppressive TAMs to orchestrate a tissue repair program promoting angiogenesis and tumor progression, a functional polarization fostered by hypoxia, which is exacerbated after extensive endothelial cell death caused by high-dose (>10Gy) radiation [92, 93]. Interestingly, while vascular death has been implicated in cures by high-dose radiation [94], recent work using an elegant spontaneous carcinogenesis model with selective deletion of ataxia-telangiectasia mutated (ATM) in either endothelial or sarcoma cells demonstrated that the increased death of ATM-deficient endothelial cells failed to improve tumor eradication by SBRT [95]. While the role of the immune system in tumor eradication was not evaluated in this study, it is intriguing to consider if TAM-mediated immunosuppression played a role in precluding tumor eradication.
Radiation also upregulates hypoxia-inducible factor-1α (HIF1-α), a key transcription factor that, in addition to promoting angiogenesis [96], induces PD-L1 expression on both tumor cells and myeloid cells in the tumor microenvironment, inhibiting T cell-mediated tumor rejection [97, 98].
Thus, the effects of radiation are complex and dependent on several variables, including the tumor type, the pre-existing tumor microenvironment, and the radiation dose used. Improved understanding of treatment protocols and combinations that strike the right balance of immune-activating versus immunosuppressive signals is critical for the optimal use of radiotherapy to generate an in situ tumor vaccine.
30.7 Revisiting Current Treatment Protocols: Lymphopenia Induced by Local Radiotherapy
An important practical consideration for the use of radiation in combination with immunotherapy is the volume of tissue that is irradiated or “field size.” As discussed in the beginning of the chapter, traditional techniques use large fields and often include draining lymph nodes and adjacent bones, with hematopoietic marrow. Despite these efforts to include and treat microscopic disease at the margins of a tumor, in many cases such as advanced head and neck cancer, locoregional recurrence rates remain ~40%, even with high total doses, large CTV, and significant short- and long-term toxicity. Traditional radiobiological paradigms explain these failures as inadequacy of radiation dose or the margins. However, with the emerging role of radiation in activating the immune system to control cancer cells, this paradigm is being reevaluated. One key adverse effect of large field radiation is killing of lymphocytes that reside or are circulating (and exposed when the beam is on, during radiation delivery) within the irradiated region. Since lymphocytes are very radiosensitive, their exposure reduces the availability of cells that might participate in tumor rejection.
Radiation-induced lymphopenia has been modeled in glioblastoma multiforme [99], which also often incorporates large fields to the brain, an organ highly perfused. Severe and persistent treatment-related lymphopenia occurs in 40% of patients undergoing standard treatment. Importantly, lymphopenia predicted for decreased survival: hazard ratio for death attributable to 2-month CD4 count below 200 = 1.66 (p = 0.03) [100].
In a modeling effort to predict this effect on blood, the mean radiation dose to circulating lymphocytes was calculated to be 2.2 Gy, and after 30 fractions, 99% of circulating blood had received ≥0.5 Gy, which was dependent on the margins used [99]. To examine the impact of margins on lymphopenia in the setting of locally advanced pancreatic cancer, total lymphocyte counts were recently compared between stereotactic and standard tridimensional fields of treatment [101]. At 2 months, 46% compared to 13.6% of patients were severely lymphopenic with standard compared to stereotactic treatment. Again, higher total lymphocyte counts posttreatment were associated with improved overall survival, further indicating the detrimental effects of large treatment fields.
Irradiation of circulating T cells may have significant effects on maintenance of immunological memory, which requires continuous homeostatic turnover [102]. In addition, functional impairments in the T cell compartment have been reported. T cells harvested during radiotherapy from peripheral blood of patients and restimulated in vitro showed reduced proliferation, with impairment still detectable 4 weeks after completion of radiotherapy [103]. Persistent lymphopenia post-radiotherapy has been linked to a persistent failure of homeostatic cytokine responses [104]. Thus, considerations for the volume of tissue irradiated and the number of fractions used are likely to be critical for successful combinations of radiotherapy and immunotherapy. Preliminary evidence suggests that classical regimens of fractionated radiotherapy protracted over 4–8 weeks of daily fractionation to large target volumes should be avoided.
30.8 Exploiting Radiation Effects to Improve Responses to Immunotherapy
The priming and effector phases of the antitumor immune response are modulated by multiple checkpoints that need to be countered to generate a clinically effective tumor rejection. Immunotherapies currently approved or under investigation target one or a few of the blocks but in many cases have failed to achieve detectable clinical responses [105]. The ability of radiation to affect many processes at both the priming and effector phase makes it an especially attractive partner in combination with different immunotherapies. Examples of preclinical combination studies are listed in Table 30.1. While Table 30.1 is not exhaustive of the large available literature, the data illustrated provides strong support for the hypothesis that radiation can work in concert with different strategies to improve tumor control. Interestingly, the radiation doses tested in different studies vary widely, but in only few studies, different doses and fractionation schema have been tested side by side, precluding in most cases any conclusion about the most effective dose. Moreover, only a few studies carefully examine the effects of the combinations on abscopal tumors. While in-field control is a necessary condition for abscopal responses, enhanced in-field responses don’t necessarily translate to abscopal effects. Thus, preclinical data should be used with caution to infer which radiation dose(s) should be tested in clinical studies [4].
Table 30.1
Radiotherapy improves responses to multiple immunotherapies in preclinical tumor models
Main step of antitumor immune response targeted | Agent | Main effect | Radiotherapy tested | Reference(s) |
|---|---|---|---|---|
Tumor antigen presentation | Flt3-ligand | Growth factor for DCs, increased systemic availability of DCs | 60 Gy X 1 2 Gy and 6 Gy X1 | [106] [15] |
Exogenous DC s.c., i.v. Exogenous DC s.c., i.t. | Increase local and systemic availability of DCs Increased availability of DCs in irradiated tumor | 10 Gy X 3–5 8.5 Gy X 5 15 Gy X 1 | [62] [107] [108] | |
CpG s.c. peri-tumorally and i.t. | TLR9 agonist, DCs activation | 10–55 Gy X 1 | [109] | |
Imiquimod, topical R848, i.v. | TLR7 agonist, local DCs activation TLR7 agonist, systemic DCs activation | 8 Gy X 3 10 Gy X 1 | [78] [110] | |
ECI301 (CCL3 variant) i.v. 2′3′-cGAMP, i.t. Vaccinia and avipox recombinants expressing CEA and T cell costimulatory molecules Autologous tumor cell vaccine expressing GM-CSF | Recruitment of DCs, T cells, NK cells STING agonist, IFN-I production Generation of tumor antigen-specific T cells | 6 Gy X 1 20 Gy X 1 8 Gy X 1 4 Gy X 2 | [111] [68] [46] [45] | |
T cell priming and activation | Anti-CTLA-4 antibody, i.p. | Immune checkpoint inhibitor | 12 Gy X 1–2 6 Gy X 5, 8 Gy X 3, 20 Gy x 1 | [73] [16] |
Anti-CD137 antibody, i.v or i.p. | Costimulatory receptor agonist | 5 Gy X 1, 10 Gy X 1, 15 Gy X 1 4 Gy X 2 | [112] [76] | |
Anti-OX40 antibody, i.p. IL-2, i.t. NHS-IL-2 (fusion of antibody to necrotic DNA with modified IL-2), i.v. TGFβ neutralizing antibody, i.p. | Costimulatory receptor agonist T cell growth factor Modified T cell growth factor, targeted to tumor Blocks TGFβ immunosuppressive effects on DC and T cells | 20 Gy X 3 2 Gy X 10 3.6 Gy X 5 6 Gy X 5 | [113] [114] [115] [37] | |
Killing of cancer cells | Anti-PD-1 antibody, i.p. Anti-PDL-1 antibody, i.p. Adoptively transferred T cells | Immune checkpoint inhibitor Blocks PD-1 ligand on tumor cells/infiltrating myeloid cells Activated tumor antigen-specific CD8 T cells | 12 Gy X 1 10 Gy X 1 12 Gy X 1, 20 Gy X 1 2 Gy X 5 8 Gy X 1 10 Gy X 1 2 Gy X 1 | [77] [116] [74] [75] [53] [47] [44] |
CSF1R inhibitor | Reduced post-RT recruitment of MDSC and TAM | 3 Gy X 5 5 Gy X 1 | [90] [117] |
30.9 Clinical Translation of Combinations of Radiotherapy and Immunotherapy: A Work in Progress
Some of the combinations of radiation and immunotherapy that were shown to have additive or synergistic effects in preclinical models have been tested in the clinic. Pioneering studies tested strategies to increase antigen-presenting cell function and/or activation with the use of growth factors (granulocyte-macrophage colony-stimulating factor, GM-CSF) or TLR agonists (CpG) achieving close to 30% abscopal responses in solid tumors and lymphoma, respectively [27, 30].
Recent groundbreaking success of checkpoint inhibitors targeting CTLA-4 and PD-1 signaling has led to rapid FDA approval of multiple agents (nivolumab, pembrolizumab, ipilimumab, and atezolizumab) for metastatic melanoma, lung cancer, renal cell carcinoma, urothelial cancer, head and neck cancer, and Hodgkin lymphoma. However, the response rates of single-agent checkpoint inhibitors remain ~15–30%, depending on the disease, and resistance develops in the majority of cases [118]. Thus, radiation is being investigated in combination with these agents for the ability to overcome resistance and potential to generate a more robust and prolonged response in close to a hundred trials [119]. Most of these studies are in the phase I/II setting; however the combination of radiation and PD1 inhibition is currently being tested in phase III studies in non-small cell lung cancer (NCT02768558), head and neck cancer (NCT03040999), and glioblastoma (NCT02617589).
A few studies have reported results. In a phase III trial, 799 patients with castrate-resistant metastatic prostate cancer that had progressed after docetaxel treatment were randomized to receive a single 8 Gy radiation dose to a bone lesion followed by ipilimumab (10 mg/kg) or placebo every 3 weeks for up to 4 doses. The endpoint of improved survival in ipilimumab versus placebo group was not achieved [120]. However, within the group of patients with better prognostic characteristics and no visceral metastases, ipilimumab improved median survival, suggesting that selection of patients with less advanced disease and possibly a better immune function may be important [121].
Results of two phase I trials testing radiotherapy and ipilimumab in metastatic melanoma have been reported, both enrolling 22 patients. In both studies, the toxicities were similar to what is expected with ipilimumab alone. In the first study, 18% of patients had partial response (best outcome), and 18% showed stable disease in nonirradiated lesions [122], an outcome not significantly different from what would be expected with ipilimumab alone [123]. In the second study, 27.3% of patients had objective responses, including three patients with complete response and three with partial response, and 22.7% had stable disease [124]. Complete responses are rarely seen in melanoma patients treated with ipilimumab alone, suggesting that the addition of radiation can indeed enhance responses. Interestingly, the two studies differed in the sequencing of radiation and ipilimumab: ipilimumab was given 3–5 days after radiation in the first study and within 5 days before radiation in the second study, suggesting the possibility that sequencing is critical. Administration of immune checkpoint inhibitors before or during radiation was also shown to be more effective than administration after radiation in preclinical studies [75, 125].
Results have also been reported for the phase I of a phase I/II trial (NCT02239900) testing SBRT with ipilimumab in advanced solid tumors, including NSCLC (n = 8), colorectal carcinoma (n = 4), sarcoma (n = 3), and renal cell carcinoma (n = 3), with a total of 31 patients evaluable for abscopal responses. SBRT was given at 12.5 Gy X 4 fractions or 6 Gy X 10 fractions to a metastasis in either the liver or the lungs concurrently with ipilimumab or sequentially (1 week after the second dose). Clinical benefit (partial response or stable disease lasting 6 months or longer in nonirradiated lesions) was seen in 23% of patients. This result is encouraging in tumors known to be largely unresponsive to ipilimumab alone but too early to determine whether the radiation dose, site of irradiation, or sequencing with ipilimumab make a difference [126]. Increased peripheral blood CD8+ T cells and CD8/CD4 T cell ratio was associated with clinical benefit. Interestingly, greater expression of some T cell activation markers was seen in patients receiving radiation to the liver than lung, but this remains to be validated.
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


