Fig. 9.1
Effect of pyrazole or LPS or LPS plus pyrazole on serum ALT or AST levels (A) and liver histopathology (B). Panels in (B) refer to (A) saline; (B) pyrazole-treated; (C) LPS-treated; (D) and (E) LPS plus pyrazole-treated. Arrows show necrotic foci with inflammatory cell infiltration. **p < 0.01 compared to all other groups. Either the JNK inhibitor SP600125 or the p38 MAPK inhibitor prevents the LPS plus pyrazole elevation of ALT and AST
To assess whether oxidative stress occurs after the various treatments, malondialdehyde (MDA) levels as a reflection of lipid peroxidation, were assayed. Whereas pyrazole alone or LPS alone did not elevate TBAR levels over those found with saline controls, the combination of LPS plus pyrazole increased MDA levels about 65 % (p < 0.05 compared to the other three groups, Fig. 9.2A). Levels of 3 nitrotyrosine protein adducts as a marker for oxidized nitrated protein formation were determined by slot blot analysis. Low levels of 3-NT adducts were found in saline control livers. Treatment with either LPS alone or pyrazole alone slightly elevated 3-NT protein adduct levels; however, striking increases in protein carbonyls were found in the combined LPS plus pyrazole group (Fig. 9.2B). Thus oxidative/nitrosative stress was elevated in livers from the LPS plus pyrazole-treated mice.
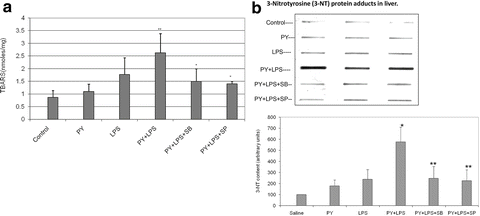
Fig. 9.2
Pyrazole plus LPS increases oxidative stress. Mice were treated with either saline or pyrazole alone or LPS alone or LPS plus pyrazole. (A) Lipid peroxidation was assayed as formation of thiobarbituric acid-reactive substances (TBARS). (B) Formation of three nitrotyrosine protein adducts was assayed by slot blot and results expressed as arbitrary units. The inhibitors of JNK (SP) and p38 MAPK (SB) lower the potentiated increase in oxidative stress produced by pyrazole plus LPS
CYP2E1 catalytic activity (oxidation of p-nitrophenol to p-nitrocatechol) was increased about twofold by either the pyrazole alone or the pyrazole plus LPS treatments. LPS alone slightly but not significantly decreased CYP2E1 activity. Levels of CYP2E1 protein, measured by immunoblot analysis, showed similar trends, being increased about twofold by pyrazole or pyrazole plus LPS treatments. This enhanced liver injury is associated with elevated levels of CYP2E1 and increased oxidative/nitrosative stress generated by the combination of LPS plus CYP2E1.
9.6 Pyrazole Potentiates TNFα Toxicity [53, 54]
Since TNFα levels are elevated after LPS administration and TNFα plays an important role in the effects of LPS, we determined if pyrazole treatment to induce CYP2E1 potentiates TNFα toxicity as it did LPS toxicity. Basically, the same approaches described above for LPS were used, with injection of TNFα (50 μg/kg body wt) replacing the LPS treatment. Figure 9.3A shows that ALT and AST levels were low in the saline control mice and in the pyrazole-treated mice challenged with saline. Treatment of control mice with TNFα elevated transaminase levels by about 2–3-fold. Treatment of the pyrazole mice with TNFα elevated transaminase levels more than 3-fold over the TNFα-saline control treated mice (Fig. 9.3A). Liver sections were stained with H&E for morphological evaluation. The saline and TNFα treated mice showed normal liver morphology. Liver from pyrazole-treated mice showed some vacuolar degeneration. Liver from the TNFα plus pyrazole-treated mice showed several necrotic loci (arrows), typical pathology morphology changes including nuclear pyknosis, karyorrhexis, and karyolysis were observed (Fig. 9.3B). The treatment with pyrazole did not significantly alter the levels of thiobarbituric acid-reactive substrates (TBARS) in the total liver extract or the mitochondria (Fig. 9.4A). TNFα treatment of control mice elevated levels of TBARS about 2–3-fold. TBARS in the homogenates and the mitochondria were further elevated when TNFα was administered to the pyrazole-treated mice. Highest liver and mitochondrial TBARs levels were observed in the pyrazole plus TNFα treated mice. Liver GSH levels were similar in the saline, pyrazole-treated, and TNFα-treated mice but were decreased about 40 % in the liver extracts from the pyrazole plus TNFα treated mice (Fig. 9.4B). GSH levels were lowered 40 % in the liver mitochondria from the pyrazole plus TNFα-treated mice compared to the TNFα alone treated mice. These results suggest that the combined pyrazole plus TNFα treatment produces elevated oxidative stress in the liver compared to TNFα alone or pyrazole alone, and that mitochondrial oxidative stress may occur in livers of the pyrazole plus TNFα-treated mice.
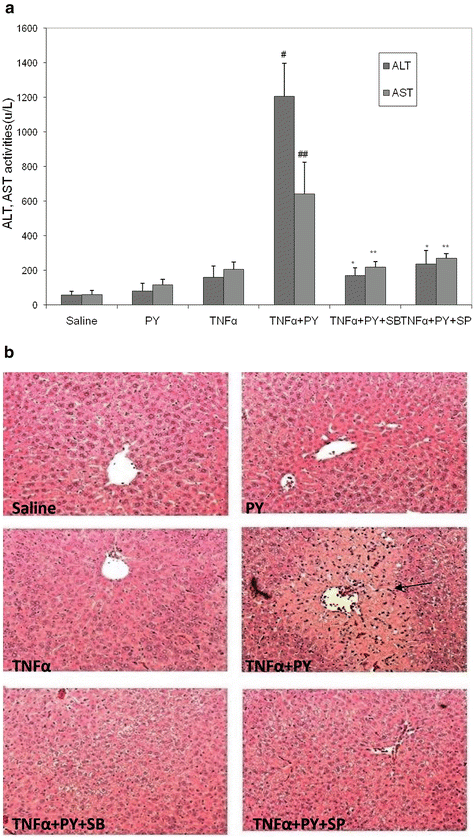
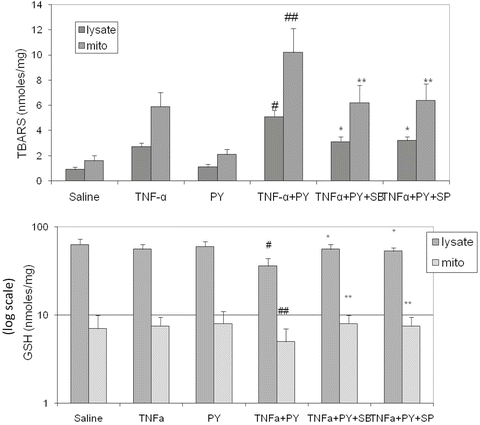
Fig. 9.3
Pyrazole potentiates TNFα hepatotoxicity and in mice and inhibitors of JNK or p38 MAPK block the increase in toxicity. Mice were treated with either saline or pyrazole alone or TNFα alone or pyrazole plus TNFα followed by assays for serum ALT/AST (A) or histopathology (B) (arrows show necrotic zones)
Fig. 9.4
Pyrazole potentiates TNFα-induced oxidative stress and inhibitors of JNK or p38 MAPK block the increase. Liver homogenates and mitochondria were isolated from mice treated as described in the legend to Fig. 9.3. Lipid peroxidation (A) and levels of GSH (B, note the log scale) were determined in the homogenates and the mitochondrial fraction
As expected, CYP2E1 activity and the content of CYP2E1 were elevated 2–3-fold by pyrazole or by pyrazole plus TNFα treatment, over the saline or TNFα alone treated mice. However, induction of CYP2E1 alone by pyrazole is not sufficient to induce liver injury; rather, a second “hit” e.g., TNFα is required.
9.7 Role of CYP2E1 in Pyrazole Potentiation of LPS and TNFα Toxicity
To validate the role of CYP2E1 in the potentiation of LPS toxicity by pyrazole, experiments with chlormethiazole (CMZ) an inhibitor of CYP2E1 and with CYP2E1 knockout mice were carried out [51]. C57BL/6 mice were injected intraperitoneally with pyrazole, 150 mg/kg body wt once a day for 2 days or 0.9 % saline. After an overnight fast, LPS, 4 mg/kg body wt or saline was injected IP. CMZ was injected in some mice at a concentration of 50 mg/kg body wt 15 h before and 30 min after the LPS treatment. Mice were killed 24 h after LPS or saline injection. In other experiments, CYP2E1 knockout mice, kindly provided by Dr. Frank Gonzalez, NCI, NIH (20) and their genetic background SV129 controls were treated with pyrazole and LPS as above. Administration of CMZ to the LPS plus pyrazole-treated mice decreased the elevated ALT and AST levels by about 55 and 65 %, respectively (Fig. 9.5A). Pathological evaluation showed large necrotic areas in the livers from the LPS plus pyrazole-treated mice, but only small necrotic foci were observed after treatment with CMZ (Fig. 9.5B). The treatment with CMZ also lowered the elevated oxidative/nitrosative stress produced by the LPS plus pyrazole treatment as only weak signals for formation of 4-HNE adducts and 3-NT adducts were found after the CMZ treatment (Fig. 9.5C). The pyrazole plus LPS treatment produced a twofold increase in CYP2E1 catalytic activity, which was prevented after the administration of CMZ. Thus, CMZ blocked the elevation of CYP2E1 in the LPS plus pyrazole-treated mice, and this was associated with a decline in oxidative/nitrosative stress and blunting of liver injury.
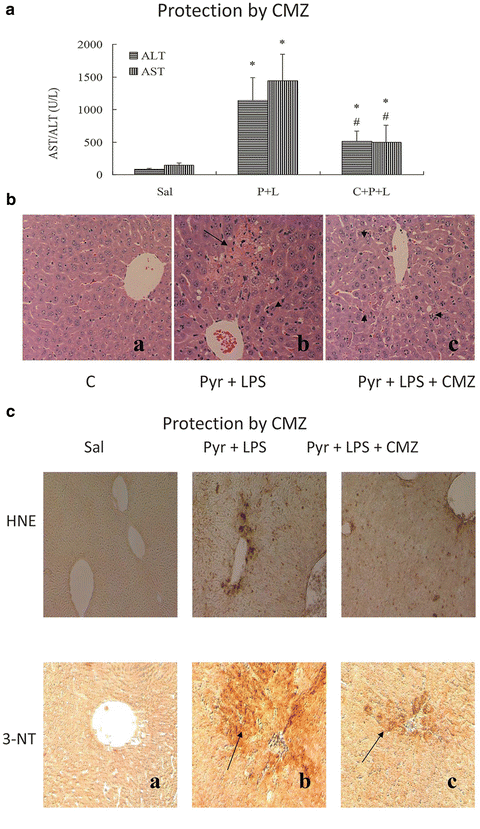
Fig. 9.5
The CYP2E1 inhibitor, chlormethiazole (CMZ) protects against LPS plus pyrazole toxicity and oxidative/nitrosative stress in mice. Sal Saline-treated; P + L pyrazole plus LPS-treated, C + P + L CMZ plus pyrazole plus LPS-treated. (A) ALT/AST levels, (B) histopathology; (C) 4-HNE adducts and 3 NT adducts
CYP2E1 knockout or wild type control SV129 mice were treated with LPS plus pyrazole. As with C57Bl/6 mice, liver injury was observed in the wild type SV129 mice treated with LPS plus pyrazole, but not mice treated with LPS alone or pyrazole alone. Serum ALT and AST levels were about 50 % lower in LPS plus pyrazole-treated CYP2E1 knockout mice as compared to wild type mice (Fig. 9.6A). Pathological evaluation showed large necrotic areas and widespread necrotic foci in wild type mice whereas almost normal histology was found in the LPS plus pyrazole-treated CYP2E1 knockout mice (Fig.9.6B). Positive TUNEL staining was also significantly lower in the CYP2E1 null mice compared to wild type mice (Fig. 9.6C). Immunoblots confirmed the absence of CYP2E1 protein in the knockout mice while strong signals from CYP2E1 were detected in immunoblots of the wild type mice (Fig. 9.6D). Thus, in both rats and mice, the CYP2E1 inducer pyrazole potentiates LPS-induced liver injury. This potentiation is associated with elevated oxidative/nitrosative stress and is blocked by the CYP2E1 inhibitor CMZ, and blunted in CYP2E1 knockout mice. We hypothesize that CYP2E1-mediated oxidative stress may synergize with LPS-generated oxidative stress in this model to produce liver injury.
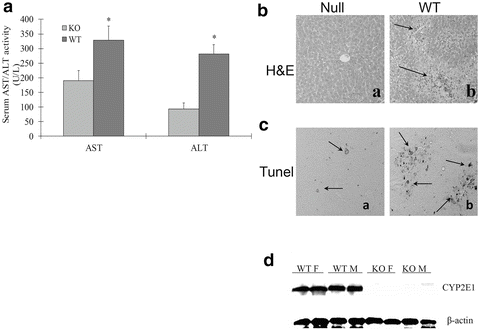
Fig. 9.6
LPS plus pyrazole toxicity is lowered in CYP2E1 knockout mice. (A) ALT/AST levels: (B) histopathology: (a) LPS plus pyrazole-treated CYP2E1 knockout mice; (b) LPS plus pyrazole-treated wild type control. (C) TUNEL staining for DNA fragmentation: (a) the CYP2E1 null mice and (b) wild type mice. (D) Immunoblot for CYP2E1 levels in wild type female or male mice (WTF, WTM) or male and female CYP2E1 knockout mice (KOM, KOF)
Similar results were found with the TNFα plus pyrazole potentiated toxicity model. Large increases in ALT and AST levels were found after TNFα administration to pyrazole-treated SV129 wild type mice. TNFα treatment of pyrazole-treated CYP2E1 knockout mice did not elevate transaminase levels. Similarly, TBAR levels in liver homogenates and isolated mitochondria were not elevated in the TNFα plus pyrazole-treated CYP2E1 knockout mice but were increased in the wild type mice. Normal liver pathology was observed after pyrazole plus TNFα treatment of CYP2E1 knockout mice. The failure of TNFα to induce liver injury in pyrazole-treated CYP2E1 knockout mice supports a critical role for CYP2E1 in the potentiated injury observed in the wild type mice.
9.8 Mitochondrial Dysfunction
Alcohol can cause mitochondrial dysfunction [55, 56]. We hypothesized that mitochondria are an eventual target for developing liver injury induced by TNFα when CYP2E1 is elevated by pyrazole. Initiation of a mitochondrial permeability transition was determined by assessing mitochondrial swelling in the absence and presence of 100 μM calcium. Succinate (10 mM) was the respiratory substrate. As shown in Fig. 9.7A, in the absence of calcium, swelling (decrease in absorbance at 540 nm) was low with all mitochondrial preparations although there was some basal swelling with the mitochondria from the pyrazole plus TNFα-treated mice. The addition of 100 μM calcium caused a low rate of swelling in the saline or TNFα alone mitochondria; swelling was somewhat elevated in the pyrazole alone mitochondria. Swelling was very rapid without any lag phase with the mitochondria from the pyrazole plus TNFα-treated mice (Fig. 9.7A). Importantly, this rapid swelling was blocked by Cyclosporine A (CsA) (2 μM), a classic inhibitor of the mitochondrial permeability transition.
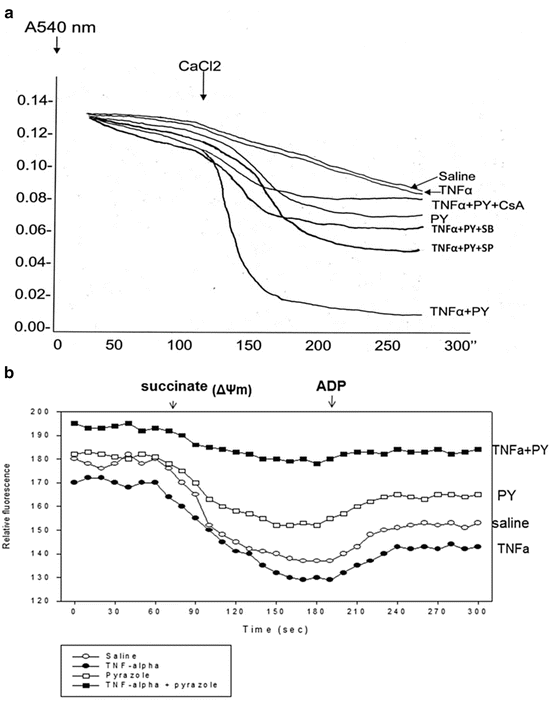
Fig. 9.7
Treatment with TNFα plus pyrazole causes mitochondrial injury as reflected by (A) increased mitochondrial swelling (decreased absorbance at 540 nm) or (B) decreased mitochondrial membrane potential as assayed by succinate-dependent decline in rhodamine 123 fluorescence. The increased swelling produced by TNFα plus pyrazole is prevented by cyclosporine (CsA), an inhibitor of the mitochondrial permeability transition as well as by the inhibitors of JNK and p38 MAPK
The electrochemical potential of the proton gradient generated across the mitochondrial membrane (ΔΨ) was assessed by monitoring fluorescence quenching of rhodamine 123. Addition of 10 mM succinate at one minute caused a decrease in fluorescence reflective of a high ΔΨ corresponding to state 4 of respiration (Fig. 9.7B). The decline in fluorescence averaged about 40 arbitrary units per minute with mitochondria from the saline or TNFα alone treated mice and 30 arbitrary units per minute with mitochondria from the pyrazole-treated mice. However, the decline in fluorescence was only about 14 arbitrary units with mitochondria from the TNFα plus pyrazole-treated mice. Addition of ADP at 3 min caused an enhancement of fluorescence which corresponds to state 3 respiration as part of the proton motive force is utilized to synthesize ATP. This enhancement of fluorescence averaged 15, 14, 12, and 4 arbitrary units per minute for mitochondria from the saline, TNFα alone, pyrazole alone, and TNFα plus pyrazole-treated mice, respectively. Taken as a whole, these initial data suggest a small decline in ΔΨ in mitochondria from the pyrazole-treated mice and a more pronounced decline in mitochondria from the pyrazole plus TNFα-treated mice.
9.9 Cyclosporine A Prevents Pyrazole Plus LPS-Induced Liver Injury [57]
We evaluated whether CsA, an inhibitor of the mitochondrial permeability transition, could protect against the TNFα plus pyrazole-induced liver injury. Such an experiment could validate that mitochondrial dysfunction is a key downstream target in this injury. Male C57BL/6 mice were treated with saline, pyrazole, LPS, or pyrazole plus LPS plus corn oil or pyrazole plus LPS plus 1 dose of CsA (100 mg/kg body wt, dissolved in corn oil). Serum ALT and AST levels were elevated in the PY + LPS + corn oil group compared to the other three groups. CsA treatment attenuated this increase in transaminases (Fig. 9.8A). H&E staining of liver tissue showed that the PY + LPS + corn oil treatment induced extensive liver zonal necrosis and that the CsA treatment prevented this (Fig. 9.8B). Mitochondrial swelling was increased in mitochondria isolated from the PY + LPS + corn oil treated mice compared to mitochondria from the saline + corn oil mice. The in vivo treatment with CsA prevented this increase in mitochondrial swelling, which likely explains the protection against LPS plus pyrazole-induced liver injury. The LPS plus pyrazole elevation of 4-HNE and 3-NT protein adducts were also decreased by CsA, suggesting that mitochondrial dysfunction plays an important role in the increase in oxidative/nitrosative stress.
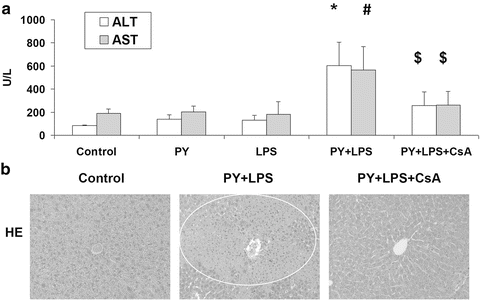
Fig. 9.8
In vivo administration of the mitochondrial permeability transition inhibitor, cyclosporine A (CsA) lowers LPS plus pyrazole-induced increase in transaminase levels (A) and liver injury as detected by H&E staining (B)
9.10 Activation of MAP Kinases
Mitogen-activated protein kinases (MAPKs) are serine-threonine kinases that mediate intracellular signaling associated with a variety of cellular activities including cell proliferation, differentiation, survival, death, and transformation. The mammalian MAPK family consists of extracellular signal-regulated kinase (ERK), p38 MAPK, and c-Jun NH2-terminal kinase (JNK) [58]. The MAPK signaling cascade consists of three distinct members of the protein kinase family, including MAP kinase (MAPK), MAPK kinase (MAPKK), and MAPKK kinase (MAPKKK). MAPKKK phosphorylates and thereby activates MAPKK, and the activated form of MAPKK in turn phosphorylates and activates MAPK. Activated MAPK may translocate to the cell nucleus and regulate the activities of transcription factors and thereby control gene expression [59]. In either in vivo or in vitro models of ALD, an increase of gene expression and activation of the MAPK pathway was found [60–62].
JNK or p38 MAPK have been shown to play important roles in several models of liver injury, including CYP2E1-dependent toxicity [49, 50, 63–65]. We evaluated possible activation of MAP kinases in our pyrazole/LPS or pyrazole/TNFα hepatotoxicity models. LPS treatment alone did not cause significant JNK activation (Fig. 9.9A) or p38 MAPK activation (Fig. 9.9B) as reflected by the low p-JNK and pp38 MAPK levels relative to total JNK and p38 MAPK levels. Similar low ratios were found for the saline or the pyrazole alone treated mice (Fig. 9.9a, B). However, both JNK and p38 MAPK were activated in livers of the pyrazole plus LPS-treated mice. A similar activation of JNK and p38 MAPK was also observed after pyrazole plus TNFα but not in mice treated with TNFα or pyrazole alone [53]. ERK was not altered by TNFα alone or pyrazole plus TNFα treatment. To evaluate the significance of these changes in MAPK activation, the effect of SP600125, an inhibitor of JNK, and SB203580, an inhibitor of p38MAPK on the hepatotoxicity was determined. The LPS (Fig. 9.1) or TNFα (Fig. 9.3) plus pyrazole elevation of transaminases was blunted by administration of SP600125 (15 mg/kg) or SB203580 (15 mg/kg) (Figs. 9.1 and 9.3). The MAPK inhibitors also lowered the necrosis, the mitochondrial dysfunction (Fig. 9.7) and partially blocked the increased oxidative stress (Figs. 9.2 and 9.4) produced by the pyrazole plus LPS/TNFα treatment, but had no effect on CYP2E1 activity or protein levels. These results suggest the CYP2E1 elevation of LPS/TNFα liver injury and oxidative stress is MAPK dependent. The activation of JNK in the pyrazole plus TNFα group was blocked by SP600125 but not SB203580 whereas the activation of p38 MAPK was blocked by SB203580 but not SP600125, validating their specificity.
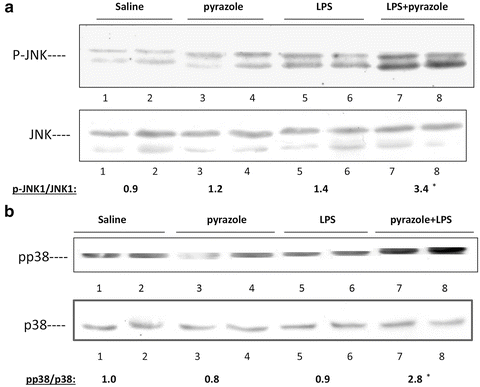
Fig. 9.9
LPS plus pyrazole treatment activates JNK (A) and p38 MAPK (B). The p-JNK/JNK and pp38 MAPK/p38 MAPK ratios are shown below the blots
9.11 Activation of ASK-1 and Downstream MAP Kinase Kinases
The upstream mediators of JNK and p38 MAPK activation were not identified in these previous studies. Apoptosis signal-regulating kinase 1 (ASK-1) is a member of the MAP3K family which is responsive to stress-induced cell damage. Activation of ASK-1 can determine cell fate by regulation of both the MKK4/MKK7-JNK and the MKK3/MKK6-p38 MAPK signaling cascades [66]. ASK-1 is activated by oxidative stress, ER stress, and inflammatory cytokines such as TNFα [67]. In resting cells, ASK-1 forms an inactive complex with reduced thioredoxin (Trx). Under conditions of stress by TNFα or ROS, ASK-1 dissociates from Trx and becomes activated [68]. Oxidation of Trx by ROS causes dissociation of ASK-1 from the oxidized Trx which switches the inactive form of ASK-1 to the active kinase. The Trx-ASK complex is thought to be a redox sensor, which functions as a molecular switch turning the cellular redox state into a MAP kinase signaling pathway [69]. Activated ASK-1 then promotes activation (phosphorylation) of the downstream MAPKK, MKK4/MKK7 which can activate JNK, and MKK3/MKK6 which can activate p38 MAPK [70, 71]. We evaluated whether CYP2E1 plus TNFα induced ROS promote release of ASK-1 from the Trx-ASK1 complex and activate ASK-1 followed by the phosphorylation of MKK4/MKK7 and/or MKK3/MKK6 which subsequently regulate the phosphorylation of JNK and p38 MAPK and contribute to the liver injury [72].
Treatment with TNFα for 4–12 h or pretreatment with PY alone did not activate ASK-1 (Fig. 9.10a). TNFα plus PY treatment activated ASK-1 about threefold compared with the 0 h control at 4 h after TNFα treatment. Activation of ASK-1 decreased at 8 and 12 h. Immunoprecipitation of Trx1 showed that ASK-1 was bound to Trx-1 at 0 h but was released from the Trx-ASK1 complex at 4 h and remained free from binding to Trx1 at 8 and 12 h (Fig. 9.10b). No ASK-1 release from the Trx-ASK1 complex was found in TNFα alone treated mice. ASK-1 was not activated in PY plus TNFα treated CYP2E1−/− mice and no ASK-1 was released from the Trx-ASK1 complex in CYP2E1−/− mice. WT mice treated with pyrazole plus TNFα developed liver injury between 8 and 12 h after addition of the TNFα (Fig. 9.10C), Oxidative stress is a likely key factor to trigger signaling and liver injury in CYP2E1-mediated hepatotoxicity [73]. Thus, activation of ASK-1 by treatment with TNFα plus PY is associated with its release from the Trx-ASK1 complex, occurs prior to the liver injury, and requires CYP2E1.
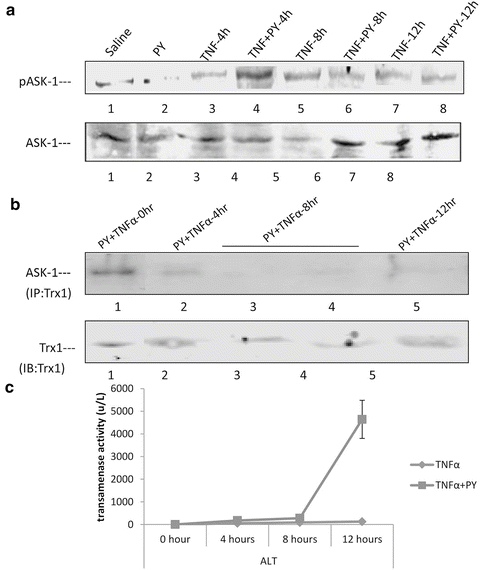
Fig. 9.10
Activation of the MAPKKK ASK-1 by pyrazole plus TNFα treatment. (A) Immunoblot showing activation (phosphorylation) of ASK-1 4 h after treatment with pyrazole plus TNFα but not by pyrazole alone or TNFα alone. (B) Dissociation of the inhibitory thioredoxin 1 (Trx1) from its complex with ASK-1 4 h after treatment with pyrazole plus TNFα. Note that Trx1 is bound to ASK-1 initially (0 h) but not 4–12 h after treatment with pyrazole plus TNFα. Liver homogenatenates were immunoprecipitated with Trx1 antibody and then the immunoprecipitates were probed for ASK-1 by Western blot. (C) Time course for liver injury produced by pyrazole plus TNF α. Note injury occurs after 8 h, a time point when ASK-1 has already been activated
MKK4/7 and MKK3/6 are the MAPKK which activate downstream JNK or p38 MAPK, respectively [71]. They are also targets for activation by ASK-1 [69–71]. Treatment of wild type mice with PY plus TNFα activated MKK4 at 4, 8, and 12 h compared with the TNFα alone groups [72]. No activation of MKK4 was found in TNFα or TNFα + PY treated CYP2E1−/− mice [72]. MKK7 was activated only at 12 h. MKK3 was activated as early as 4 h in the TNFα plus PY treated mice while MKK6 was activated at 8 h. JNK was activated in the TNFα + PY mice at 8 and 12 h and p38 MAPK was activated at 12 h when compared with TNFα alone. In CYP2E1−/− mice, neither MKK4/7, MKK3/6, JNK, nor p38 MAPK was activated. Thus, the time course experiments suggest MKK4 may be the MAPK responsible for activation of JNK, while either MKK3 or MKK6 may be the MAPKK responsible for the activation of p38 MAPK.
Our results implicate a role for ASK-1 in CYP2E1 potentiation of TNFα-induced liver injury. Future experiments with ASK-1 knockout mice [74] would be interesting to further validate the role of ASK-1 in the PY/TNFα model. In CYP2E1−/− mice, no MAPKK was activated at any observation time point. TNFα alone did not significantly activate the MAPKK in wild type or CYP2E1−/− mice. The activation of MKK4 and MKK3/6 (4–8 h) occurs prior to the onset of liver injury (8–12 h). We hypothesize that TNFα alone-or CYP2E1 alone-generated ROS stress is not sufficient to trigger the dissociation of ASK-1 from the Trx-ASK complex. The CYP2E1 sensitization of TNFα induced liver injury may occur through a synergistic effect with TNFα to produce an enhanced ROS stress consistent with the so call “Two Hit” hypothesis.
9.12 Effect of N-Acetylcysteine
We evaluated [54] the effect of N-acetylcysteine (NAC), a general antioxidant and a precursor of GSH, on the potentiation of TNFα toxicity by pyrazole as a proof of principle that oxidative stress plays an important role in the overall liver injury. C57BL/6 mice were treated with pyrazole for 2 days and then challenged with either saline or TNFα. Some mice in each group were also treated with 150 mg/kg NAC on the second day of treatment with pyrazole and on day 3 prior to the challenge with TNFα. The elevation in ALT and AST and the necrosis caused by the pyrazole plus TNFα treatment were lowered by NAC. The increase in TBARs produced by pyrazole plus TNFα and the decline in liver GSH were both prevented by NAC. Treatment with NAC had no effect on CYP2E1 protein levels or CYP2E1 catalytic activity. The activation of JNK or p38 MAPK by the pyrazole plus TNFα treatment was blocked by NAC. These results with NAC suggest that elevated oxidative stress is central to the activation of JNK and p38 MAPK and to the liver injury produced by treatment with pyrazole plus TNFα.
9.13 Hepatotoxicty by CYP2E1 Plus TNFα Occurs in JNK2 but not JNK1 Knockout Mice
The hepatotoxicity produced by pyrazole plus LPS/TNF-α was prevented by SP600125, an inhibitor of JNK (Figs. 9.1 and 9.3). JNK is encoded for by three genes, each of which is alternatively spliced to yield α and β forms of both a 54 and 46 kDa protein. In hepatocytes, only two of the genes, JNK1 and JNK2 are expressed [75]. Mice deficient in either JNK1 or JNK2 are viable but double knockouts are embryonic lethal suggesting some redundant functions [75]. JNK has been implicated in hepatic injury produced by TNF-α, ischemia-reperfusion, hepatitis virus, bile acids, alcohols and acetaminophen [64, 65, 76–78]. Recent studies have evaluated whether JNK1 or JNK2 play the more predominant role in potentiation of liver injury. In fibroblasts, JNK1 but not JNK2 appears to be essential for TNF-α-induced apoptosis [79]. However, liver injury produced by either LPS/d-galactosamine or TNF-α/d-galacosamine was the same in WT and JNK1 KO mice but lower in JNK2 KO mice [80]. JNK2 promoted the development of steatohepatitis in mice fed a methionine choline-deficient diet [77]. Singh et al. [81] reported that JNK1 KO mice fed a high fat diet did not gain weight or develop steatohepatitis as did the WT and JNK2 KO mice. JNK2 was found to be predominant in acetaminophen toxicity [65]. 6-Hydroxydopamine-induced apoptosis in PC12 cells was JNK2 but not JNK1 dependent [82]. It appears that depending on the toxin and cell type, either JNK1 or JNK2 or both play the major role in cell injury. We evaluated whether JNK1 or JNK2 or both play critical roles in the potentiation of TNF-α-induced hepatotoxicity and oxidative stress by pyrazole [83].
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcohol and HCV: Implications for Liver Cancer
Alcohol and HCV: Implications for Liver Cancer
 Alcohol Metabolism by Oral Streptococci and Interaction with Human Papillomavirus Leads to Malignant Transformation of Oral Keratinocytes
Alcohol Metabolism by Oral Streptococci and Interaction with Human Papillomavirus Leads to Malignant Transformation of Oral Keratinocytes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


