Pancreatic neuroendocrine tumors (pancNETs) are rare neoplasms that comprise 2% to 4% of all clinically detected pancreatic tumors. They are usually indolent, and their malignant potential is often underestimated. The management of this disease poses a challenge because of the heterogeneous clinical presentation and varying degree of aggressiveness. Treatment decisions for this clinical entity are still patient- and/or physician-specific. Optimal clinical management of pancNETs requires a multidisciplinary approach. The only potentially curative treatment option, especially in the early stage disease, remains surgical resection; however, as many as 75% of patients present with advanced disease (nodal and/or distant metastases).
Key points
- •
Pancreatic neuroendocrine tumors (pancNETs) comprise 2% to 4% of all detected pancreatic tumors; they can be indolent, and yet their malignant potential is often underestimated.
- •
The management of this disease poses a challenge because of the heterogeneous clinical presentation and varying degree of aggressiveness.
- •
Surgical therapy remains the most efficient approach and offers the longest lasting benefits for patients with pancNETs.
- •
Clinical management of pancNETs involves benefits substantially from a multidisciplinary approach.
Introduction
Pancreatic neuroendocrine tumors (pancNETs) are uncommon tumors with an estimated incidence of 1 to 1.5 per 100,000 and a prevalence of 35 per 100,000 in the United States. They originate from the embryonic endodermal cells that give rise to the islets of Langerhans. These cells are specialized cells that produce, store, and secrete peptides and biogenic amines and were formerly known as the amine precursor uptake and decarboxylation cells or APUD cells.
PancNETs comprise 2% to 4% of all pancreatic neoplasms, and peak incidence is found between the sixth to the eighth decades. Data from the SEER (Surveillance, Epidemiology, and End Results) registry have shown an increase in incidence from 0.17 in 1970 to 0.43 in 2007, with the lowest 5-year survival compared with other gastrointestinal neuroendocrine tumors (NETs) ( Fig. 1 ).
PancNETs are often classified as functional (F) or nonfunctional (NF) based on the presence or absence of clinically evident hormone production. NF tumors are more common than functional tumors. Functional tumors secrete one or more biologically active peptides, which may result in systemic clinical symptoms ( Table 1 ). NF is likely a misnomer because these tumors often secrete various peptides, albeit in the absence of a clinical syndrome (see Table 1 ).
| pNET | Syndrome Name | Primary Location(s) | Incidence (No. of New/100,000/y) | Malignancy (%) | Hormone-Causing Syndrome |
|---|---|---|---|---|---|
| Functional pNETs | |||||
| Gastrinoma | ZES | Pancreas (30%), duodenum (60%–70%), other (5%–10%) | 0.5–1.5 | 60–90 (30–560) | Gastrin |
| Insulinoma | Insulinoma | Pancreas (100%) | 1–3 | 5–15 | Insulin |
| VIPoma | Vemer-Morrison, Pancreatic cholera, WDHA | Pancreas 85%–95%, other (neural, periganglionic, adrenal) (10%) | 0.05–0.2 | 70–90 | Vasoactive intestinal peptide |
| Glucagonoma | Glucagonoma | Pancreas (100%) | 0.01–0.1 | 60–75 | Glucagon |
| Somatostastinoma | Somatostastinoma | Pancreas (50%–60%), duodenal/jejunal (40%–50%) | <0.1%, uncommon | 40–60 | Somatostatin |
| GRFoma | GRFoma | Pancreas (30%), lung (54%), jejunal (75%), other (adrenal, foregut, retroperitoneal) (13%) | Unknown | 30–50 | Growth hormone–releasing factor |
| ACTHoma | ACTHoma | 4%–25% of all ectopic Cushing syndrome | <0.1%, uncommon | 95 | ACTH |
| PET causing carcinoid syndrome | PET causing carcinoid syndrome | Pancreas (100%) (<1% of all carcinoid syndrome) | Uncommon (<50 cases) | 60–90 | Serotonin, tachykinins |
| PET causing hypercalcemia | PTHrPoma | Pancreas (100%) | <0.1%, uncommon | >85 | PTHrP, other unknown |
| NF pNET | PPomas NF-PET | Pancreas (100%) | 1–5 | 60–90 | None secrete pancreatic polypeptide (PP) (60%–85%), chromogranin A but cause no symptoms |
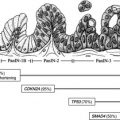
Efforts have been made to create a universal classification system of these tumors from a prognostic and therapeutic standpoint. In 2000, under the auspices of the World Health Organization (WHO), a NET classification was proposed with 3 categories: well-differentiated tumors with benign or uncertain behavior; well-differentiated carcinoma with malignant characteristics; and poorly differentiated carcinoma ( Table 2 ). This classification, updated in 2010, takes into account the anatomic location, mitotic activity, and the Ki67 proliferative index. This classification was then incorporated into the seventh edition of the American Joint Committee on Cancer staging manual and into the National Comprehensive Cancer Network (NCCN) guidelines.
| WHO 1980 | WHO 2000 | WHO 2010 |
|---|---|---|
|
|
|
a G2 NET may include WDET or WDEC of the WHO 2000 classification.
General features of pancreatic neuroendocrine tumors
There are 10 different commonly recognized pancNETs, of which 9 are associated with a clinical syndrome, including gastrinomas, insulinomas, glucagonomas, VIPomas, GRFomas, ACTHomas, somatostatinomas, pancNETs causing carcinoid syndrome, and pancNETs causing hypercalcemia. Amounts of 60% to 100% of NF pancNETs secrete various peptides such as chromogranin A, neuron-specific enolase, pancreatic polypeptide, ghrelin, neurotensin, motilin, or subunits of human chorionic gonadotropin, all of which cause no obvious clinical syndrome. Insulinomas, gastrinomas, and nonfunctioning pancNETs were previously reported as representing about a third of all pancNETS, respectively. More recent reports suggest pancNETs occur with increasing frequency, making up twice the above percentage of pancNETs, often discovered when still asymptomatic and without advanced disease.
Some pancNETs are part of 1 of 4 different inherited syndromes: multiple endocrine neoplasia type-1 (MEN-1), von Hippel-Lindau disease (VHL), von Recklinghausen disease neurofibromatosis 1, and tuberous sclerosis ( Table 3 ). These tumors frequently differ in clinical presentation, prognosis, and management from sporadic pancNETs. Of these, MEN-1 is the most important inherited pancNET because 20% to 80% of all patients with this autosomal-dominant disorder develop a clinically relevant NET. MEN-1 is found in 20% to 25% of patients with gastrinomas, 4% with insulinomas, and less than 3% with other pancNETs. Almost all patients with MEN-1 have multifocal, asymptomatic NF-pancNETs, whereas symptomatic pancNETs occur in less than 10% of this group of patients.
| Syndrome | Frequency | Location/Type of Genetic Abnormality | Altered Protein Function(s) | Frequency of PETs, % | Type of PETs (%) |
|---|---|---|---|---|---|
| MEN-1 (Wermer syndrome) | Prevalence, 1–10 per 100,000 | 11q13; encodes 610 amino acid protein (menin) | Nuclear location; exact function unclean interacts with JunD, NF-kB, SMAD signaling pathways; effects cell cycle, growth, genomic stability, and apoptosis | 80–100 (microscopic), 20–80 (clinical) | NF-PET, microscopic; > functional (20–80) |
| VHL | Prevalence, 2–3 per 100,000 | 3p25: encodes 232 amino acid protein (pVHL) | Interacts with elongins, which act as transcriptional regulators that degrade HIF, regulates cell cycle, VEGF | 10–17 | NF (>98) |
| Von Recklinghausen disease (neurofibromatosisl [NF-1]) | Prevalence, 1 per 4000–5000 | 17q11.2: encodes 2485 amino acid protein (neurofibromin) | Ras GTPase-activating activity, binds microtubules, modulates adenylate cyclase, mTor-regulates growth, cell cytoskeleton | 0–10 (uncommon) | Duodenal somatostatinomas, rare PETs |
| Tuberous sclerosis (Bourneville disease) | Prevalence, 1 per 10,000 | 9q34 (TSC1): encodes 1164 amino acid protein (hamartin); 16p13 (TSC2): encodes 1807 amino acid protein (tuberin) | Interacts with PI3K signaling pathway regulating GTPase and mTor, which play a key role in growth, energy regulation, response to hypoxia, nutrients | Uncommon | Rarely develop functional, NF-PETs |
Diagnosis of pancNETs is often delayed for months to years, given their indolent nature and relatively nonspecific symptoms, even for patients with functional tumors. Around 60% to 70% of patients have metastatic disease at presentation, most commonly involving the liver and less frequently the bones. With the exception of insulinomas, where fewer than 10% are considered malignant, pancNETs typically demonstrate malignant behavior in at least 50% of cases.
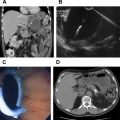
Preoperative imaging and localization are essential before considering surgical resection of these tumors. The imaging modalities most commonly used are triple-phase multidetector computed tomography (CT) scan and MRI with gadolinium contrast. PancNETs are usually hyperenhancing masses in the arterial phase of the scan (both primary and metastatic lesions) due to their hypervascular nature ( Fig. 2 ). Contrast-enhanced ultrasound is used more frequently in Europe than in the United States and may be useful in monitoring response to treatment with peptide receptor radionuclide therapy (PRRT) and locoregional ablation of liver metastases. As most pancNETs (excluding insulinomas) express a high density of somatostatin receptors (specifically subtypes 2 and 5), indium-111-labeled somatostatin receptor scintigraphy (SRS) is an effective localizing tool in this disease. There is evidence that SRS informs us of tumor receptor status, and that this may be useful for guiding therapeutic use of somatostatin analogues. Recent development of PET scanning with gallium-68-labeled somatostatin analogues (DOTATOC, DOTANOC, and DOTATATE) may be more sensitive than CT, MRI, and octreoscan in detecting NETs (100% vs 75%).
Management steps for pancNETs include establishing diagnosis, localizing tumor, assessing for underlying inherited disorder, controlling hormonal excess when present, removing and/or ablating tumor if possible and appropriate, and considering alternative forms of therapy (regional and/or medical). Depending on the situation, pancNETs can either be enucleated or removed with formal organ resection. The former approach is considered in cases of smaller tumors with presumed indolent behavior, assuming pancreatic duct disruption can be avoided. Intraoperative ultrasound (IOUS) is an integral part of resection of these tumors. If the above criteria are not met, formal resection, such as pancreaticoduodenectomy, distal pancreatectomy, and the less commonly performed central pancreatectomy, is considered.
Surgical management of localized pancreatic neuroendocrine tumors
Insulinoma
Insulinoma is the most common form of F-pancNET encountered. It can be sporadic or associated with MEN-1. It is more commonly found in women (F:M = 2:1) in the fifth or sixth decade and can present in the third decade of life in patients with MEN-1. Sporadic insulinomas are usually unifocal, but those associated with MEN-1 may present with multifocal disease. Clinical presentation is characterized by the Whipple triad constituted by hypoglycemia after a fast or exercise, neuroglycopenic symptoms, and immediate relief with oral or intravenous glucose administration. Most patients also present with hyperphagia and new-onset weight gain.
The diagnostic criteria for insulinoma are established during a 72-hour fast or until symptoms develop with a blood glucose less than 50 mg/dL, elevated insulin, C-peptide, and proinsulin, in the absence of urine or plasma sulfonylureas, and relief of symptoms after an oral glucose load ( Box 1 ); 99% of insulinomas can be detected in this manner. Approximately 80% to 90% of insulinomas are small (<2 cm), solitary, benign tumors that are equally distributed among the head, body, and tail of the pancreas. The remaining 10% occur as multiple tumors in the setting of MEN-1. Most insulinomas are less than 2 cm in diameter and are benign, with a brick-red appearance due to increased vascularity. Given their small size, preoperative localization remains a challenge. CT scanning can detect approximately two-thirds of all lesions on arterial phase imaging. MRI is typically performed as a second-line imaging modality after CT, although the sensitivity for detecting these tumors may be higher than that of a CT scan. The sensitivity of endoscopic ultrasound (EUS) varies with tumor location (92.6%, 78.9%, and 40.0% for the head, body, and tail of the pancreas, respectively) and with user experience. SRS for this type of tumor is limited because only 30% possess somatostatin type 2 receptors. Intra-arterial calcium stimulation (selective infusion of calcium into branches of the celiac axis and superior mesenteric artery) with hepatic venous sampling for insulin can be a sensitive localizing modality, but carries some risk of complications, and is typically used when the tumor or tumors are not localized with other forms of imaging ( Fig. 3 ).
Monitored fast for less than 48 hours with documented blood glucose less than 50 mg/dL with hypoglycemic symptoms
Relief of symptoms after oral glucose load
Elevated insulin level (>5–10 μU/mL)
Increased serum proinsulin level (>22 pmol)
Absence of urinary or plasma sulfonylureas
Elevated C-peptide levels
Despite this, 20% to 50% of insulinomas remain undetected at the time of surgery. Some evidence suggests that preoperative localizing studies for the primary tumor may not be necessary. This finding is based on the observation that a combination of surgical exploration and IOUS can detect more than 90% of insulinomas.
Definitive treatment of insulinomas is surgical resection; however, presurgical medical therapy is provided to alleviate symptoms, including eating small, frequent meals and using insulin antisecretagogues, such as diazoxide or octreotide, both of which work only 40% to 60% of the time. Before proceeding to surgery, the presence of MEN-1 must be excluded by testing for other components such as hyperparathyroidism and pituitary tumors. Intraoperative glucose monitoring is essential to avoid hypoglycemia, and dextrose infusion must be stopped before surgical resection to permit intraoperative glucose measurements as an indicator of biochemical cure. Many sporadic adenomas are amenable to enucleation. Surgical exploration commences by gaining access to the pancreas in the lesser sac and traversing the gastrocolic omentum. A wide kocherization of the duodenum is essential for tumor involving the pancreatic head. This wide kocherization enables palpation to be combined with IOUS, which in experienced hands can detect up to 98% of insulinomas. It also helps to detect the location of the pancreatic duct to ascertain safety during enucleation and prevent a pancreatic fistula. Once the insulinoma is enucleated, a thorough examination must be performed to evaluate for a ductal leak, in the presence of which a suture repair can be attempted along with surgical drainage ( Fig. 4 ). Rates of pancreatic fistula reported after enucleation are higher than those after a formal resection, in the range of 18% to 38%.
In MEN-1 patients, these tumors are multiple and subcentimeter and usually coalesce. If multiple tumors are found throughout the pancreas, judicious use of surgical resection must be considered because total pancreatectomy should be avoided for insulinomas in most cases. If the lesion is not found, a pancreatic biopsy specimen should be obtained to rule out nesidioblastosis (a condition characterized by diffuse β-cell proliferation), in which case a subtotal pancreatectomy can improve symptoms.
No follow-up is necessary in the case of benign insulinomas unless symptoms recur. For malignant insulinomas and those associated with MEN-1, follow-up is every 3 to 12 months with biomarkers, CT/MRI, and octeroscan/PET.
The treatment of malignant/metastatic pancNETs is discussed in a separate section.
Gastrinoma
Gastrinomas are the second most common functional islet cell tumor of the pancreas. Mean age at diagnosis is 50 years with a slight male predominance. Although slow growing in nature, more than 60% demonstrate malignant behavior. Because of their indolent growth pattern, 10-year patient survival approaches 90% even in the presence of metastatic disease. Two-thirds of all gastrinomas are sporadic, with the remainder associated with MEN-1. Individuals with gastrinomas associated with MEN-1 may have a better 20-year survival rate than individuals with sporadic gastrinomas. As with other pancNETs, gastrinomas in the setting of MEN-1 tend to be multifocal, and as many as 50% of patients have distant metastases at the time of presentation.
The clinical syndrome associated with gastrinoma (Zollinger-Ellison syndrome [ZES]) arises principally from dysregulated hypergastrinemia and subsequent luminal hyperacidity resulting in development of intractable gastrointestinal ulcers. Diarrhea, heartburn, nausea, and weight loss are the typical constellation of symptoms at presentation. With the advent and widespread use of antisecretory medications, patients may or may not present with intractable ulcer disease at the time of diagnosis. Given the nonspecificity of symptoms and relative rarity of this disorder, the mean time to diagnosis is around 5.9 years. The diagnostic criteria ( Table 4 ) include an increased basal acid output in the presence of a fasting gastrin level (FSG) >100 pg/mL or >10-fold the upper limit of normal. Proton pump inhibitors (PPIs) are stopped at least a week prior and H2 antagonists 2 days before testing. Because almost two-thirds of patients with ZES have equivocal FSG levels, secretin stimulation testing or calcium provocative testing can be useful in establishing the diagnosis. An increase of 200 pg/mL in the serum gastrin level following secretin administration is consistent with ZES. Higher serum gastrin levels are seen with pancreatic rather than duodenal primaries and in patients with tumors larger than 3 cm and/or those with liver metastases.
Related posts:
 Molecular and Genetic Basis of Pancreatic Carcinogenesis
Molecular and Genetic Basis of Pancreatic Carcinogenesis
 Role of Endoscopic Ultrasonography and Endoscopic Retrograde Cholangiopancreatography in the Clinical Assessment of Pancreatic Neoplasms
Role of Endoscopic Ultrasonography and Endoscopic Retrograde Cholangiopancreatography in the Clinical Assessment of Pancreatic Neoplasms
 Minimally Invasive Approaches to Pancreatic Surgery
Spectrum and Classification of Cystic Neoplasms of the Pancreas
Clinical Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors
State-of-the-art Imaging of Pancreatic Neuroendocrine Tumors
Minimally Invasive Approaches to Pancreatic Surgery
Spectrum and Classification of Cystic Neoplasms of the Pancreas
Clinical Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors
State-of-the-art Imaging of Pancreatic Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


