Fig. 29.1
Treg depletion mediated by low-dose chemotherapy and IL-2-/IL-2R-targeting drugs. (a) Low-dose oral metronomic chemotherapeutic agents including cyclophosphamide (CTX), paclitaxel (PTX), and fludarabine (FDB) induce a profound and selective decrease on Treg number. (b) IL-2/IL-2 receptor blockade agents: daclizumab and basiliximab, two anti-CD25 antibodies (radiolabeled or not), can invoke cell death (apoptosis) by mediating ADCC or CDC. Two CD25-directed immunotoxin antibodies are being tested in human cancers: IMITOX-25 (RFT5-dgA), a CD25 antibody RFT5 linked to deglycosylated ricin A, and LMB-2, a fusion protein composed of the Fv portion of CD25 antibody attached to a fragment of Pseudomonas exotoxin A. Due to its IL-2 component, denileukin diftitox/ONTAK is capable of binding to CD25 (high-affinity IL-2 receptor) causing its rapid endocytosis and resulting in Treg cell apoptosis
Ghiringhelli and colleagues were the first to demonstrate that a single dose of CTX (30 mg/kg, i.p.) can deplete Tregs from tumor-bearing rats compared with nontreated animals [46]. These preclinical results were further confirmed by the same group in patients with advanced solid tumors [43]. They showed a strong and selective decrease of peripheral immunosuppressive Tregs, improving the control of tumor progression. Moreover, these authors described metronomic CTX as a potent treatment for reducing tumor-induced immune tolerance before anticancer immunotherapy.
Furthermore, Yingzi Ge et al. monitored in treatment-refractory and metastatic-advanced breast cancer patients the immunological effect of metronomic CTX (50 mg/day per OS during 3 months) on circulating Treg number and function, as well as on the induction of tumor-specific T-cell responses. The authors measured a transient Treg depletion with a stable breast tumor-reactive T-cell response, which was correlated with good clinical outcomes in these patients [47].
Treg depletion by low-dose chemotherapy prior to vaccination showed a significant enhanced overall survival (OS) in patients and elicited an effective antitumor immune response, as reported in many murine tumor models and in different human cancers. For example, in a randomized phase II study using a peptide-based vaccine for renal cancer treatment, it has been shown that a single dose of CTX reduced the number of Tregs and induced specific immune responses, associated with longer OS [48].
In a recent phase I/II clinical trial, metastatic castration-resistant prostate cancer (mCRPC) patients received a long-term administration of a DC-based vaccine combined with chemotherapy drugs (metronomic CTX, docetaxel, and prednisone). A significant decrease in Tregs in the peripheral blood was observed. The long-term administration of the vaccine led to the induction and maintenance of tumor-specific T cells, but no immunological parameter was significantly correlated with better OS [49].
In contrast to these clinical outcomes, in a phase I clinical trial, 49 metastatic cancer patients (breast, lung, kidney, stomach, bladder, and prostate cancers) received a bacille Calmette–Guérin (BCG) vaccine plus CTX, to induce Treg depletion. Surprisingly, the number of peripheral Tregs was significantly increased upon treatment [50]. One possible explanation for the opposite effect of CTX in this study could be the difference between the routes of administration. In fact, in the clinical studies mentioned above, CTX was orally administrated, whereas in the latter example, patients received a single intravenous infusion of CTX.
However, other reports supported that metronomic drugs, even administrated orally, induced an increase of Treg number and suppressive activity. Koumarianou and colleagues showed not only an increase of Treg number in the peripheral blood of cancer patients but also an increase of Treg/effector T-cell (Teff) ratio and of the immunosuppressive activity of Tregs. They also found that these effects were more profound in metronomic than in standard chemotherapeutic approaches [51].
In a phase II trial, Ellebaek et al. also showed that despite using metronomic CTX in combination with a COX-2 inhibitor and a DC-based vaccine, Tregs did not decrease after the treatment in melanoma patients [52].
To date, the reasons for the contradictory effects of metronomic chemotherapy on Tregs are not clear [53]. The doses and the schedule of drug administration, the type and stage of cancer, the mode of action of the chemotherapeutic drugs, the basal status of the immune system of patients before treatment, and the choice of combinations should be taken into particular account by clinicians in the design of experimental approaches including metronomic chemotherapy drugs.
29.3 IL-2 Receptor (IL-2R)-Induced Treg Depletion
Tregs are characterized by the expression of IL-2R α chain or CD25, which is critical to their expansion [54]. IL-2R on Tregs also acts as a regulatory mechanism. High-affinity IL-2R on Tregs limits the amount of IL-2 available for Tconv, hindering their expansion and activation [1]. This is the reason why some Treg-depleting strategies use the IL-2/IL-2R axis to deplete Tregs in cancer and, therefore, restore antitumoral immunity.
29.3.1 Anti-CD25 Antibodies (e.g., Daclizumab and Basiliximab)
Daclizumab is a humanized IgG1-κ monoclonal antibody (mAb) targeting human CD25. It is well tolerated when administered and causes no severe secondary effects [55]. Initially approved by the Food and Drug Administration (FDA) in 1997 for the prevention of renal allograft rejection, it is now been tested for the depletion of Tregs (Fig. 29.1b) in several cancer clinical trials [56] (Table 29.1).
Table 29.1
CD25-targeting agents in cancer clinical trials
Treatment | Cohort | Indication | Trial phase | Start date—status | NCT ID |
|---|---|---|---|---|---|
LMB-2 an anti-Tac(Fv)-PE38 recombinant immunotoxin + peptide-based vaccine | 26 | Metastatic melanoma | Phase II | 12/2005 (completed) | NCT00295958 |
IMTOX-25 (RFT5-dgA) anti-CD25 immunotoxin | 41 | Metastatic melanoma | Phase II | 04/2006 (completed) | NCT00314093 |
Basiliximab (Simulect®) antihuman CD25 chimeric antibody + DC-based vaccine | 18 | Glioblastoma multiforme | Phase I | 03/2007 (active, not recruiting) | NCT00626483 |
Daclizumab + peptide-based vaccine + Prevnar | 11 | Metastatic breast cancer | Phase I | 11/2007 (completed) | NCT00573495 |
IMTOX-25 | 29 | Advanced cutaneous T-cell non-Hodgkin lymphoma | Phase II | 07/2008 (completed) | NCT00667017 |
LMB-2 + fludarabine + CTX | 18 | Adult T-cell leukemia | Phase II | 10/2008 (active, not recruiting) | NCT00924170 |
Daclizumab + tumor lysate-loaded DC-based vaccine or + bevacizumab | 67 | Recurrent ovarian, primary peritoneal, or fallopian tube cancer | Phase I | 05/2010 (active, not recruiting) | NCT01132014 |
Yttrium-90-labeled daclizumab + chemotherapy + auto-stem cell transplant | 6 | Advanced Hodgkin lymphoma | Phase I/II | 10/2011 (active, not recruiting) | NCT01468311 |
Yttrium Y 90 basiliximab + chemotherapy (before autologous hematopoietic cell transplantation) | 24 | Mature T-cell non-Hodgkin lymphoma | Phase I | 06/2015 (recruiting) | NCT02342782 |
The first group to investigate the effect of daclizumab on Treg depletion in cancer used a single low dose of 0.5 mg/kg, prior to a DC vaccination on metastatic melanoma patients [57]. They reported that daclizumab depleted CD4+ CD25high FoxP3+ T cells from patients’ peripheral blood but measured a clearance of daclizumab associated with the reappearance of Tregs approximately 30 days after. However, the functional T-cell-specific response to the vaccine was less effective on patients pretreated with daclizumab, compared to the group who received only the vaccine.
In another trial, Rech et al. investigated the effect of daclizumab on metastatic breast cancer patients in association with a peptide-based vaccine [58]. The authors observed that daclizumab permitted both a marked and durable depletion of Tregs in peripheral blood and an effective boosting of vaccine-induced specific T-cell responses.
Those studies demonstrated that antihuman CD25 daclizumab is capable of inducing Treg depletion in cancer patients. Treg depletion can lead to CD25− T-cell activation, and the remaining antibody in the system would likely be deleterious to newly activated T cells expressing CD25 [59]. In fact, daclizumab in vivo half-life is over 4 weeks, which could explain the lower rate of T-cell-specific response after vaccination observed in Jacobs et al. study.
Basiliximab, a chimeric antibody, is another antihuman CD25 mAb, which is assessed in cancer clinical trial for Treg depletion (Fig. 29.1b). It is currently being tested in different phase I clinical trials in glioblastoma, acute myeloid leukemia, and Hodgkin and non-Hodgkin lymphoma cancer patients (Table 29.1).
29.3.2 Denileukin Diftitox (ONTAK®)
Denileukin diftitox (DD)/ONTAK is a genetically engineered recombinant fusion protein composed of diphtheria toxin catalytic domain and the full-length human interleukin-2 (IL-2). Due to its IL-2 component, DD is capable of binding to high-affinity IL-2R causing its rapid endocytosis [60] and resulting in cell apoptosis within 40–72 h [61] (Fig. 29.1b). ONTAK has been approved by the FDA since 1999 for the treatment of patient with persistent or recurrent cutaneous T-cell lymphoma (CTCL), whose malignant cells express IL-2R α chain CD25 [62]. Since then, ONTAK has also been investigated in CD25− cancers, to deplete CD25+ Treg populations. So far, ONTAK has been used in numerous clinical trials for Treg depletion in different cancers, dosages, and regimen, all with various outcomes (Table 29.2).
Table 29.2
ONTAK (denileukin diftitox) in cancer clinical trials
Treatment | Cohort | Indication | Trial phase | Start date—status | NCT ID |
|---|---|---|---|---|---|
ONTAK denileukin diftitox + aldesleukin | 20 | Kidney cancer | Early phase I | 04/2005 (completed) | NCT00278369 |
ONTAK denileukin diftitox | 17 | Adult T-cell leukemia | Phase II | 07/2005 (completed) | NCT00117845 |
ONTAK denileukin diftitox | 15 | Advanced breast cancer | Phase I/II | 09/2005 (active, not recruiting) | NCT00425672 |
ONTAK denileukin diftitox + autologous DC-based vaccine | 24 | Adult solid tumor | Phase I | 09/2005 (completed) | NCT00128622 |
ONTAK denileukin diftitox | 69 | Stage IV melanoma | Phase II | 03/2006 (completed) | NCT00299689 |
ONTAK denileukin diftitox | 19 | Epithelial ovarian cancer, extraovarian peritoneal cancer, and fallopian tube carcinoma | Phase II | 02/2007 (completed) | NCT00880360 |
ONTAK denileukin diftitox + rituximab | 24 | Advanced non-Hodgkin lymphoma | Phase II | 04/2008 (completed) | NCT00460109 |
ONTAK denileukin diftitox | 90 | Advanced melanoma | Phase II | 06/2010 (active, not recruiting) | NCT01127451 |
ONTAK + allogeneic NK cells + rituximab + chemotherapy | 32 | Non-Hodgkin lymphoma and chronic lymphocytic leukemia | Phase II | 08/2010 (completed) | NCT01181258 |
E7777 (Eisai Inc.) improved purity ONTAK | 90 | Cutaneous T-cell lymphoma | Phase III | 05/2016 (recruiting) | NCT01871727 |
Several studies have been using ONTAK at a single dose of 18 μg/kg, in combination with DC or peptide vaccination. These trials include the treatment of carcinoembryonic antigen (CEA)-positive malignancies [63], metastatic renal cell carcinoma (RCC) [64], and metastatic melanoma [65]. Among those three studies, two reported no changes in the number of CD4+ CD25+ FoxP3+ at this dose [63, 65]. On the other hand, the RCC trial observed a transient depletion of CD4+ CD25high Tregs, with no apparent impact on other cell populations, including CD25low T cells [64].
Among the different trials about Treg depletion with ONTAK, four studies were published about metastatic melanoma. Taken together, ONTAK showed its efficacy to transiently deplete Tregs in metastatic melanoma patients at 12 μg/kg/day for 4 consecutive days [66, 67], at 5 and 18 μg/kg/day for 3 consecutive days [68], but not at the dose of 18 μg/kg for a single injection [65] nor at 9 and 18 μg/kg/day for 5 consecutive days [59]. Those results show that appropriate dosage of ONTAK is important to effectively deplete Tregs in melanoma patients as well as in other type of cancers [63, 69].
Despite the depletion of Treg populations observed in some studies, ONTAK clinical benefits have not been as strong as expected, leading to no or very limited beneficial outcomes for patients. This leads to the question: What if ONTAK has other undiscovered effects on patients’ immune system explaining its lack of efficacy? In a clinical trial, melanoma patients were treated with ONTAK to deplete Tregs before a DC-based vaccination. Interestingly, ONTAK pretreatment failed to induce tumor antigen-specific CD4+ and CD8+ T cells after numerous DC vaccinations, leading to no clinical benefits for the patients [60]. The authors demonstrated that ONTAK treatment acts not only as a Treg-depleting agent but also as a strong immunomodulator, leading to a tolerogenic DC phenotype [70, 71]. ONTAK also failed to induce apoptosis on resting Tregs, which showed increased survival [60].
Very recently, a phase III trial was launched to evaluate the safety and efficacy of E7777 (improved purity ONTAK) in persistent or recurrent CTCL (NCT01871727).
29.3.3 Anti-CD25 Immunotoxin Antibodies
Two CD25-directed immunotoxin antibodies, IMITOX-25 (RFT5-dgA), a CD25 antibody RFT5 linked to deglycosylated ricin A (after internalization, dgA induces a cell death via protein synthesis inhibition), and LMB-2, a fusion protein composed of the Fv portion of CD25 antibody attached to a fragment of Pseudomonas exotoxin A (this immunotoxin is known to induce caspase-mediated apoptosis) (Fig. 29.1b), have been developed to reduce anti-CD25 in vivo half-life (<4 h) and have been tested in clinical trials [72–74] (Table 29.1). Also, a transient depletion of CD25+ Tregs among the patients were noted, the CD25− Treg subset survived, and the depletion of CD25+ Tregs was not sufficient to increase the immune response to peptide vaccination.
In summary, daclizumab, ONTAK, and other CD25+ cell-depleting agents have shown, in some cases, great potential to deplete CD25+ Tregs in cancer patients. However, the ability of these agents to target all CD25+ cells could explain why in some trials, those molecules failed to increase vaccine-induced specific response, as CD25 marker is not specific for Tregs [75]. The persistence of CD25int/low Tregs in those studies also shows the need to develop more Treg-specific therapies or to combine them with other Treg-depleting agents to target a larger population of Tregs.
29.3.4 Emerging New Anti-CD25 Antibody Therapies
Another pitfall of anti-CD25 targeting is the systemic depletion of Tregs leading to profound disruption of peripheral homeostasis and severe side effects. An exciting new generation of anti-CD25 antibodies aims to selectively deplete intratumoral Tregs.
Recently, Sato et al. associated an anti-CD25-F(ab’)2 with a photoactivable silicon phthalocyanine dye (IRDye 700DX) to selectively deplete TI Tregs on tumor-bearing mice [76]. After injection of the photoactivable antibody, local exposure of near-infrared (NIR) light on the tumor leads to the activation of the IRDye 700DX, and therefore induces the depletion of Tregs within the TME, without damaging adjacent cells nor NIR-unexposed CD25+ systemic Tregs. After intravenous injection of the labeled antibody and exposure to NIR light, they reported a depletion of 85% of CD4+ CD25+ Tregs at the tumor site within 30 min. They also reported a rapid CD8+ T cells, NK cells, as well as antigen-presenting cells activation after NIR treatment. This local Treg depletion was followed by tumor regression and prolonged survival of the mouse after only one treatment.
FDA-approved human CD25 antibodies such as daclizumab and basiliximab reduce the barrier of translating this new treatment into clinical trial. Moreover, a phase I human study using the anti-epidermal growth factor receptor (EGFR) conjugated with the IRDye 700DX is currently being evaluated for inoperable head and neck cancer (NCT02422679), making these NIR photoimmunotherapy strategies suitable for the treatment of cancer patient in clinical trial.
29.4 Blocking Treg Cell Trafficking into Tumors
29.4.1 CCL22–CCR4 Pathway Blockade
The C-C motif chemokine receptor 4 and C-C motif chemokine ligand 22 (CCR4–CCL22) pathway represent a dominant mechanism responsible for intratumoral Treg recruitment, as described in multiple tumor types [28]. Indeed, Treg trafficking and infiltration into different tumor types appear to be dependent on the expression of CCR4 ligands (CCL17 and CCL22) produced by tumor cells or infiltrating macrophages [32] (Fig. 29.2a). Furthermore, CCR4 is highly expressed by FoxP3high CD25high CD45RA− cells, designated as effector Tregs (eTregs) [77], which are predominant among TI FoxP3+ T cells [78].
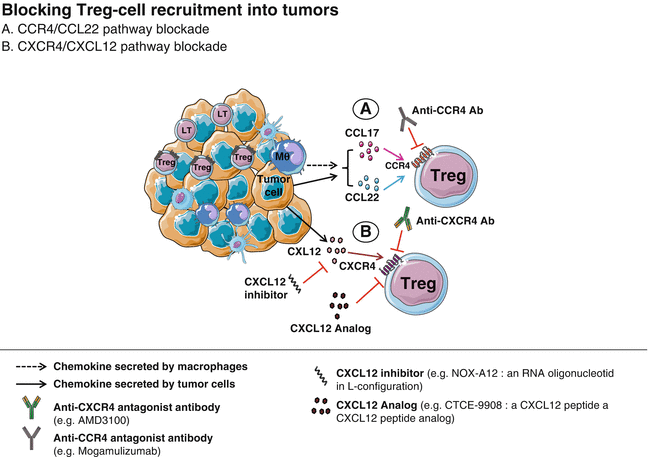
Fig. 29.2
Blocking Treg cell recruitment into tumors. Tregs are recruited into the tumor in response to chemokines secreted by malignant cells and innate immune cells (e.g., macrophages). CCL17/CCL22–CCR4 (a) and CXCL12–CXCR4 (b) are two key chemokine–chemokine receptor pathways, targeted by specific antagonist monoclonal antibodies and chemokine inhibitors and analogs (which competitively binds to the chemokine receptor). Blocking this axis may impede tumoral homing of Tregs and promote tumor growth regression
In a CTCL mouse model, Ito et al. used an anti-CCR4 antagonist mAb and showed a significant depletion of TI Tregs mediated by NK-induced antibody-dependent cell-mediated cytotoxicity (ADCC) [79]. Our group has also shown that inhibition of the CCR4 expressing Treg population by means of a CCR4 antagonist was sufficient to break immune tolerance to spontaneous mammary tumors, suggesting a major role for CCR4 in the immunosuppressive activity of Tregs [80]. All together, these observations provide a rationale for the development in clinical trials of CCR4 and CCL22 antagonists in order to block Treg cell trafficking into tumors.
So far, CCR4 antagonists have been mainly evaluated in patients with T-cell lymphoma (adult T-cell leukemia–lymphoma (ATLL), peripheral T-cell lymphoma (PTCL), and CTCL, whose tumor cells also express CCR4 at their surface) [81, 82] (Table 29.3).
Table 29.3
CCR4-targeting antibody in clinic
Treatment (company) | Cohort | Indication | Trial phase | Start date—status | NCT ID |
|---|---|---|---|---|---|
Mogamulizumab (Kyowa Hakko Kirin Co.) KW-0761 a defucosylated humanized anti-CCR4 antibody | 16 | Adult T-cell leukemia and lymphoma (ATL), adult peripheral T-cell lymphoma (PTCL) | Phase I | 02/2007 (completed) | NCT00355472 |
Mogamulizumab | 42 | PTCL or cutaneous T-cell lymphoma (CTCL) | Phase I/II | 05/2009 (completed) | NCT00888927 |
Mogamulizumab vs. vorinostat | 372 | Previously treated CTCL | Phase III | 11/2012 (active, not recruiting) | NCT01728805 |
Mogamulizumab | 72 | Advanced solid tumors | Phase I/II | 10/2014 (recruiting) | NCT02281409 |
Mogamulizumab ± MEDI4736 ± tremelimumab | 108 | Advanced solid tumors | Phase I | 11/2014 (recruiting) | NCT02301130 |
Mogamulizumab + docetaxel | 13 | Previously treated non-small cell lung cancer | Phase I | 01/2015 (completed) | NCT02358473 |
Mogamulizumab + PF-05082566 | 70 | Advanced solid tumors | Phase Ib | 05/2015 (recruiting) | NCT02444793 |
Mogamulizumab + nivolumab | 188 | Advanced solid tumors | Phase I/II | 12/2015 (recruiting) | NCT02705105 |
KHK2455 IDO-1 inhibitor ± mogamulizumab | 50 | Solid tumors | Phase I | 08/2016 (recruiting) | NCT02867007 |
Mogamulizumab (KW-0761) is a defucosylated, humanized IgG1 mAb targeting CCR4 (Fig. 29.2a), engineered to exert potent ADCC. It showed promising therapeutic potential in phase I and II clinical trials in treating CTCL which is associated with poor prognosis [81, 83, 84].
The initial phase I/II multicenter study evaluated the efficacy of mogamulizumab (dose escalation study) as a monotherapy, in pretreated patients with PTCL or CTCL. The anti-CCR4 mAb depleted efficiently the circulating CCR4+ eTregs, even at the lowest dose of 0.1 mg/kg, and restored antigen-specific cytotoxic T lymphocyte (CTL) responses [85, 86]. Recently, the same group launched a phase III trial, comparing the progression-free survival of mogamulizumab versus vorinostat (histone deacetylases inhibitor) in the treatment of CTCL (NCT01728805).
Sun et al. showed that monocyte chemoattractant protein-1 (MCP-1), an endogenous CCR4-binding ligand, was specifically upregulated in the head and neck squamous cell carcinoma (HNSCC) patients, compared to the other CCR4-binding ligands. Using a CCR4 antagonist that blocks eTreg cell recruitment induced through MCP-1/CCR4 signaling, the authors observed an inhibition of tumor growth and prolonged survival [78].
Since 2014, the mogamulizumab efficacy is evaluated in advanced or metastatic solid cancer patients. Most of these trials are using mogamulizumab in combination with other immunotherapies including anti-PD-1, anti-PD-L1 and anti-CTLA-4, anti-4-1BB antibodies, and indoleamine 2,3-dioxygenase (IDO) inhibitor (Table 29.3).
29.4.2 CXCR4–CXCL12 Pathway Blockade
The CXC chemokine ligand 12 (CXCL12), also called stromal cell-derived factor-1 (SDF-1), secreted by bone marrow, lymph node, and inflammatory cells is another chemokine responsible for the trafficking of Tregs expressing CXCR4, the receptor of CXCL12 [33, 87].
The CXCL12–CXCR4 pathway is known to be widely involved in tumor cell progression, angiogenesis, and metastasis in a number of human cancers [88, 89] including melanoma [90], ovarian [91], breast [33], small cell lung [92, 93], and gastric [94] cancers. Therefore, this axis is associated with metastasis induction [95, 96] and poor clinical outcome [33, 97], making it an attractive target for therapeutics that can block the CXCL12/CXCR4 interaction or inhibit downstream intracellular signaling.
Multiple agents are currently being developed to target the CXCL12 pathway in cancer, including AMD3100, a highly specific CXCR4 antagonist [98], also known as plerixafor; CTCE-9908, a CXCL12 analog which competitively binds to CXCR4; and a CXCL12 inhibitor, Spiegelmer/NOX-A12 (Fig. 29.2b). Among these drugs, AMD3100 and CTCE-9908 are approved for clinical use in patients with leukemias and osteosarcoma, respectively [99].
Recent studies have demonstrated the potential involvement of CXCL12–CXCR4 pathway activation in tumor resistance to conventional chemotherapies [100–102] and anti-angiogenic treatments [103]. Therefore, emergent combinatory approaches involving CXCL12 pathway blockade are being developed in order to face these drug resistance [103].
However, the impact of CXCL12–CXCR4 pathway blockade on Treg cell populations has not been fully investigated in human and requires further investigations.
29.5 Anti-angiogenic Therapies Targeting Regulatory T Cells
Vascular endothelial growth factor-A (VEGF-A) has a central role in tumor-induced immunosuppression, especially in the accumulation of Tregs [104]. VEGF-A can block DC maturation [105] or enhance myeloid-derived suppressor cell proportion in tumor-bearing hosts [106]. These two cell types can be involved in the proliferation of Tregs or the conversion of Tconv to Tregs. Moreover VEGF-R2, a VEGF-A receptor, is expressed on Tregs in tumor-bearing mice [107] and in human tumors [108] suggesting a direct role of VEGF-A on Tregs. Indeed, tumor-derived VEGF-A was shown to directly induce the proliferation of Tregs in tumor-bearing mice and in metastatic cancer patients [107].
Anti-angiogenic molecules targeting the VEGF-A/VEGF-R2 pathway are routinely used to treat many cancer patients (e.g., metastatic colorectal cancer, metastatic renal cell cancer, advanced non-small cell lung cancer, ovarian cancer). The administration of sunitinib, a tyrosine kinase inhibitor that targets VEGF-R, PDGFR, c-kit, and Flt3, decreases the proportion of Tregs in metastatic renal cell cancer (mRCC) patients [109]. This decrease has been associated with a better OS in another cohort of mRCC [110].
The negative impact of anti-angiogenic molecules on Tregs has been confirmed with another multi-target tyrosine kinase inhibitor which is sorafenib (VEGF-R, PDGFR, c-kit, Raf kinase, RET inhibitor) in mRCC patients and in hepatocellular cancer patients [111].
Bevacizumab, a humanized anti-VEGF-A mAb, also decreases the proportion and number of Tregs in tumor-bearing mice and in metastatic colorectal cancer patients [107]. Thus, VEGF-A/VEGF-R2 blockade can restore steady-state Treg proportion but can also modulate other escape mechanisms induced by the tumor (myeloid-derived suppressor cell (MDSC) induction, expression of inhibitory checkpoints on CD8+ T cells, etc.).
Based on these properties, we have shown that VEGF-A antibody can synergize with immunotherapy and especially with checkpoint inhibitors in a mouse model of colorectal cancer [112].
The association of anti-angiogenic molecules with immunotherapies is currently evaluated in different cancer locations (metastatic melanoma NCT02400385, renal cell carcinoma NCT02348008, non-small cell lung cancer NCT02039674).
29.6 Immune Checkpoint Blockade Therapy (Anti-CTLA-4 Antibodies)
CTLA-4, a co-inhibitory receptor, is transiently expressed on activated cytotoxic T lymphocytes and acts as an inhibitory molecule to reduce immune response, IL-2 production, and cell cycle [113, 114]. This negative feedback prevents lymphoproliferative and autoimmune responses in different mouse models [115–118].
In cancer, CTLA-4 expression on Teff induces hyporesponsiveness or anergy against malignant cells [119]. This characteristic of CTLA-4 has led to the development of numerous clinical trials using anti-CTLA-4 antibodies such as ipilimumab (IgG1) and tremelimumab (IgG2), two full human mAbs [120, 121]. Ipilimumab was approved by the FDA in 2011 and is currently a first-line treatment option for patients with advanced melanoma. Tremelimumab was approved by the FDA in 2015 for patients with malignant mesothelioma.
However CTLA-4 is also constitutively expressed by Tregs, which plays an important role in their regulatory function [122]. This feature led the scientific community to investigate potential effects of this therapy on Tregs, both in preclinical and clinical studies.
Kavanagh and colleagues investigated the effect of different dosage of ipilimumab on metastatic prostate cancer [113]. They observed that low dose of 1.5 mg/kg or more induced a dose-dependent increase of peripheral CD4+ FoxP3+ functional Tregs. Similar results were also found by others, showing that anti-CTLA-4 therapies can induce Treg proliferation in periphery [123–125]. However, the immune profile of the peripheral blood does not necessarily reflect what occurs in the TME.
Indeed, two preclinical studies have shown that anti-CTLA-4 mAbs induce a reduction of TI Tregs, associated with tumor regression. This CTLA-4-mediated Treg depletion was dependent on the anti-CTLA-4 isotype [126] and the Fcγ receptor (FcγR) expressing cells, present in the TME [127]. Similar results were found in human studies, where ipilimumab was able to mediate, through NK cells, an ADCC of CTLA-4+ TI Tregs (Fig. 29.3a) isolated from head and neck squamous cell carcinoma patients [128]. In melanoma patients also, ipilimumab induced ADCC mediated by FcγRIII+ nonclassical monocytes [129].
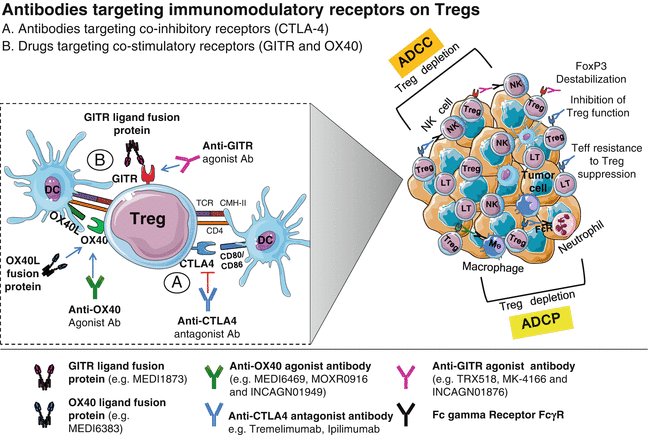
Fig. 29.3
Antibodies targeting immunomodulatory receptors on Tregs. (a) Anti-CTLA-4 mAbs may induce a reduction of TI Tregs, associated with tumor regression. Engagement of ipilimumab, an IgG1 mAb, with CTLA-4 on Tregs is able to mediate an ADCC of CTLA-4+ TI Tregs, while CTLA-4 blockade by tremelimumab, an IgG2 mAb, inhibits Treg suppressive functions and induces Teff resistance to Treg suppression. (b) Engagement of costimulatory receptors (GITR and OX40) expressed on Tregs by agonist antibodies or by their respective ligand fusion protein bearing an IgG1 Fc antibody that can also bind to activating Fc gamma receptor (on NK cells, neutrophils, and macrophages) leads to Treg depletion via ADCC or ADCP. Anti-GITR antibodies can also induce loss of Treg stability through FoxP3 destabilization
Tremelimumab, an IgG2 mAb, is not likely to do ADCC and induce subsequent TI Treg depletion. In phase I and II studies, tremelimumab showed some clinical outcomes in metastatic melanoma patients [130]. However, in a phase III study (NCT00257205), tremelimumab failed to obtain statistically significant survival advantage compared to conventional chemotherapy in first-line treatment of metastatic melanoma patients. Based on these results, the clinical trial has been halted in April 2008 [131].
On the other hand, Ménard et al. showed in advanced melanoma patients that tremelimumab was able to restore circulating effector memory T-cell proliferation and that this population became transiently resistant to Treg immunosuppressive function [119] (Fig. 29.3a).
In summary, although CTLA-4 mAbs have shown promising results in clinical trials, little is known about their effects on Treg cell populations. A better understanding of their mechanisms of action would be of great interest to develop even more potent therapies and maybe limit severe immune-related adverse events observed in various trials [120, 130, 132, 133].
29.7 Agonist Antibodies Affecting Treg Immunosuppressive Activity
Another alternative therapeutic strategy to boost antitumoral immunity is targeting costimulatory molecules involved in Treg cell modulation. We will address here two major costimulatory tumor necrosis factor (TNF) receptor superfamily members: GITR and OX40.
29.7.1 Agonist Anti-GITR Antibody
GITR, a cell surface costimulatory receptor, is highly and constitutively expressed by Tregs [11, 12], whereas naive and memory Tconv express it at low levels [134]. However, on conventional CD4+ and CD8+ T cells, GITR expression is also enhanced following activation and then declines 1–3 days later [135]. Thus, GITR signaling and function are context and cell type dependent [136].
Under suboptimal TCR stimulation, the activation of GITR has opposite effect on Tregs and conventional CD4+ and CD8+ T cells. The use of agonist anti-GITR antibodies such as DTA-1 [12], recombinant GITR ligand (GITRL), or GITRL transfectants (Fig. 29.3b) abrogates Treg cell-mediated suppression exerted on Teff cells, while it enhances the expansion and the cytokine production of CD8+ T cells [137]. This observation has been shown both in vitro [138] and in vivo in murine tumor models [139–144]. In these preclinical studies, GITR stimulation has been reported to increase Teff-to-Treg ratio [145], leading to a beneficial antitumoral effect and tumor regression in tumor-bearing mice.
Moreover, Coe et al. showed in mice that DTA-1-mediated Treg depletion was more marked in tumors than in tumor-draining lymph nodes [140], as TI Tregs express much higher levels of GITR than circulating Tregs [146]. This means that GITR could be used as a target for selective depletion or inhibition of CD4+ FoxP3+ GITRhigh Tregs without interfering with systemic maintenance of self-tolerance.
The in vivo activity of GITR-targeting agents is complex and may rely on several suggested mechanisms (Fig. 29.3b), including (1) FcγR-mediated depletion of Tregs [139], (2) activation-induced apoptosis through Fas–FasL signaling [147], (3) inhibition of Treg suppressive functions and induction of Teff resistance to Treg suppression [148], and (4) loss of Treg stability through FoxP3 destabilization [149].
Very recently, a novel hexameric GITRL, MEDI1873, based on a fusion protein bearing a human IgG1 Fc domain, has been developed by MedImmune. The assessment of MEDI1873 in vitro and in a CT26 tumor model (a murine colorectal cancer model) revealed an increased binding to FcγRs resulting in the depletion of TI Tregs, likely through Fc-mediated effector functions [150]. MEDI1873 is currently assessed in a phase I clinical study (NCT02583165) in patients with solid tumors.
In light of the preclinical results, a number of drugs, including traditional GITR-targeting antibodies and the GITRL protein fusion, have already entered [136] or have been selected for clinical trials that should be launched soon (Table 29.4).
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment




Table 29.4
GITR-targeting drugs in ongoing trial
Treatment (company) | Cohort | Indication | Trial phase | Start date—status | NCT ID |
|---|---|---|---|---|---|
TRX518 (GITR, Inc.) humanized nondepleting mAb | 40 | Malignant melanoma | Phase I | 10/2010 (recruiting) | NCT01239134 |
MK-4166 (Merck) humanized mAb ± pembrolizumab | 94 | Solid tumors | Phase I | 06/2014 (recruiting) | NCT02132754 |
BMS-986156 (Bristol–Myers Squibb) ± nivolumab | 260 | Solid tumors | Phase I/ IIa | 10/2015 (recruiting) | NCT02598960 |
MK-1248 (Merck) ± pembrolizumab | 96 | Solid tumors | Phase I | 11/2015 (recruiting) | NCT02553499 |
MEDI1873 (MedImmune LLC) hexameric GITRL protein/human IgG1 | 47 | Solid tumors | Phase I | 11/2015 (recruiting) | NCT02583165 |
TRX518 | 44 | Solid tumors | Phase I | 12/2015 (recruiting) | NCT02628574 |
INCAGN01876 (Incyte/Agenus) humanized IgG1 mAb | 146 | Solid tumors | Phase I/II | 04/2016 (recruiting) | NCT02697591 |
GWN323 (Novartis Pharma) humanized IgG1 mAb ± PDR001 | 264 | Solid tumors | Phase I/Ia | 07/2016 (recruiting) | NCT02740270 |
A non-exhaustive list of ongoing clinical trials monitoring GITR-targeting drugs in cancer patients. The choice of the samples was made as described in Table 29.1. All clinical trials can be found in https://clinicaltrials.gov/
Related posts:
Peptide-Based Therapeutic Cancer Vaccines
The Human Tumor Microenvironment
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
