C6. Storage disorders in the neonate and childhood
The clinical symptoms include pancytopenia, hepatosplenomegaly and lymphadenopathy. The central nervous system may also be affected.
α-Mannosidosis
α-Mannosidosis is an oligosaccharide storage disease characterised by deficiency of the enzyme α-mannosidase. There are two phenotypes: the infantile phenotype, which presents in the first 12 months of life and has a short survival, and the juvenile/adult phenotype, presenting later in life and having a longer survival.
The clinical features include skeletal changes, coarse facial features and mental retardation. Hepatosplenomegaly is present in the early stage of the disease. Abnormal chemotaxis of the neutrophils leads to recurrent bacterial infections.
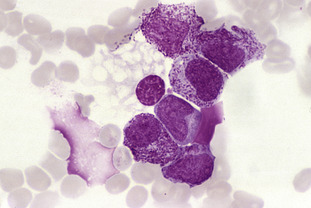
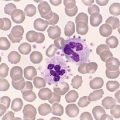
The circulating lymphocytes have vacuolated cytoplasm and the bone marrow is packed with foamy macrophages. The lymphocyte vacuoles show periodic acid Schiff (PAS) positivity.
Increased levels of mannose-containing oligosaccharides are found in the tissues as well as in the urine. Refer to Fig C6-1.
Mucopolysaccharidoses
Mucopolysaccharide storage disorders lead to morphological changes in white cells in both the peripheral blood and the bone marrow. Mucopolysaccharides are complex carbohydrates found in various types of connective tissue, including cartilage and bone. The presence of excessive amounts of mucopolysaccharide leads to coarse facial features, skeletal dysplasia and limitation of joint movement (Hurler syndrome).
Mucopolysaccharidosis results from a lysosomal enzyme deficiency.
Hurler syndrome (Gasser lymphocytes)
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


