Fig. 1
Representative images showing the different acini structures generated from immortalized normal human breast epithelial MCF10A, breast cancer MCF7 and BCSCs isolated from MCF7 population that survived a course of FR. DLG and HER2 indicate the polarity of cells in the acini
1.3 Tumor Heterogeneity and CSC-Mediated Tumor Repopulation Under Radiotherapy
Tumor heterogeneity was suspected as early as three hundred years ago when the original microscope was invented (Zellmer and Zhang 2014). As a major tumor response to therapeutic ionizing radiation, repopulation of tumor cells has been defined in experiments and clinic practice (Heppner and Miller 1989). Radiation induced repopulation of tumor cells is recognized as one of the “R”s in radiation oncology, which includes repopulation, redistribution, re-oxygenation, and repair. Such repopulation during therapy has been implicated as an important feature in tumor recurrence after radiotherapy (Kim and Tannock 2005). The pioneering work by Dewey’s group using long-term observations of irradiated cells via computerized video time-lapse analyses revealed the heterogeneity and different cell fates among irradiated cells (Endlich et al. 2000), demonstrating the heterogeneous IR response in a supposedly homogeneous cell population. Data from our lab also demonstrated that a small fraction of breast cancer MCF7 cells were able survive a course of chronic fractionated radiation (FR) and showed an enhanced profile of genes regulating cell cycle and DNA repair (Li et al. 2001; Guo et al. 2003). This radioresistant breast cancer population was found to be enriched with specific breast cancer stem cells expressing HER2 (Duru et al. 2012). Increased tumorgenicity of CSCs with specific surface markers was first studied in acute myeloid leukemia (Bonnet and Dick 1997).
A CSC is thus defined as a specific tumor cell that has stem-cell like properties including the capacity to self-renew and to generate the heterogeneous lineages of cancer cells that comprise the tumor. A key feature of CSC theory is that only a small subset of tumor cells has the ability to proliferate in an uncontrolled manner which challenges the traditional concept that each tumor cell is able to grow to a new tumor. Al-Hajj et al. demonstrated that BCSCs (CD44+/CD24−/low) are more tumorigenic; as few as 100 cells with this phenotype are able to form tumors in mice, while millions of cells without this feature cannot (Al-Hajj et al. 2003). Phillips et al. used the CD44+/CD24−/low BCSCs isolated from breast cancer cells to show that BCSCs can be propagated as mammospheres and enriched after radiation (Phillips et al. 2006). Using xenogeneic tumors treated with chemotherapy, Dylla et al. identified the repopulation of colorectal CSCs with the marker of CD44+ESA+ (Dylla et al. 2008; Dalerba et al. 2007). Firat et al. (Firat et al. 2011) further identified a delayed cell death associated with mitotic catastrophe in irradiated GSCs. These results together with other reports demonstrate that the surviving cancer cells under RT are enriched with CSCs and may contribute to tumor repopulation observed in the clinic. During chemo- and/or radio-therapy, the most resistant CSCs would be selected and continue to sustain the tumor. In clinical studies, the proportion of putative CSCs in a residual tumor has been shown to increase following cytotoxic chemotherapy (Findlay et al. 2014; McClements et al. 2013; Vaz et al. 2014; Duru et al. 2014). These findings shed light on a new conceptual paradigm of how CSCs or tumor-initiating cells contribute to radiation response. Identification of CSC-associated radioresistance needs to be further evaluated in clinical studies.
1.4 GSCs in Brain Tumor Radiotherapy
Glioblastoma multiforme (GBM) is a highly malignant primary brain tumor with a median survival time of approximately 14 months from time of initial diagnosis (Stupp et al. 2009), and radiotherapy has been the major modality for control of this tumor (Brandes et al. 2009; Balasubramaniam et al. 2007; Dhermain 2014). GSCs identified in human GBM showed infinite self-renewal and can differentiate into different cells types such as neurons and glia (Galli et al. 2004; Singh et al. 2004). GSCs also express NSC markers such as CD133, Sox2 and Nestin (Singh et al. 2004). Evidence suggests that GSCs are responsible for the aggressive tumor growth and radiotherapy resistance, although the precise mechanism has yet to be elucidated (Stiles and Rowitch 2008). Not surprisingly, Bao et al. (2006) reported that DNA damage repair capacity was enhanced in isolated GSCs. Using a spontaneous murine glioma model, Chen et al. identified that when temozolomide treatment is discontinued, the first cell population to undergo proliferation and lead to tumor regrowth is the nestin-positive GSCs (Chen et al. 2012). GSCs were also found to be enriched in recurrent gliomas. GSCs isolated from recurrent tumors form more aggressive invasive tumors in athymic mice than GSCs isolated from primary tumors derived from the same patient (Huang et al. 2008), an important result indicating the repopulation of GSCs in recurrent and metastatic tumors. In brief, the radioresistant phenotype of GBM is linked with the following factors seen in GSCs after FR: repopulation, enhancement of DNA repair (Bao et al. 2006), reprogramming of metabolism (Rampazzo et al. 2013), inhibition of apoptosis (Pareja et al. 2014; Zanotto-Filho et al. 2012; Rahaman et al. 2002), and upregulation of pro-survival factors (e.g., Akt and Mcl-1) (Choi et al. 2014; Bruntz et al. 2014). Interference of these pro-surviving signaling pathways is potential approaches to enhance the RT response, especially when recurrent GBM is treated by RT.
1.5 Profiling Radioresistant Biomarkers
IR-responsive gene expression profiles were thoroughly investigated by Amundson and Fornace and their colleagues (Amundson et al. 1999a, b; Amundson and Fornace 2001). Specific proteomics was first investigated by Dritschilo’s group in radioresistant and radiosensitive head and neck squamous carcinoma cell lines profiled using two-dimensional polyacrylamide gel electrophoresis followed by computer-assisted quantitative analysis (Ramsamooj et al. 1992). These results provide evidence supporting the fact that differential protein expression is associated with cellular radioresistance or radiosensitivity. To identify the dynamic gene expression profile, i.e., the gene expression pattern of RT-surviving cancer cells, the author’s group reported a unique gene expression profile of breast cancer MCF7 cells that survived a clinical regimen of fractionated RT (MCF7+FIR) (Li et al. 2001; Guo et al. 2003; Xia et al. 2004; Fukuda et al. 2004). This preliminary gene profiling of RT-surviving cancer cells demonstrated a cluster of genes involved in DNA repair and cell cycle regulation (Li et al. 2001). Ogawa et al. (2006) further identified a cluster of pro-survival genes in radioresistant pancreatic cancer cell lines that survived FR. Although many gene profiling studies have compared radioresistant and sensitive cancer cells (Fukuda et al. 2004; Ogawa et al. 2006; Kitahara et al. 2002; Guo et al. 2005; Lee et al. 2010), to elucidate the mechanisms causing tumor cell survival after RT, further investigation on dynamic gene and protein expression patterns in surviving cancer cells and recurrent tumors will be highly informative. Using MRM-based targeted proteomics profiling, global kinome signatures of the radioresistant MCF7/C6, a cloned cell line from MCF7+FIR population has recently been reported. Of 120 kinases studied, kinases involved in cell cycle progression including CHK1, CDK1, CDK2, and the catalytic subunit of DNA-dependent protein kinase are overexpressed and hyperactivated (Guo et al. 2015). To detect the protein expression pattern of CSCs in surviving cancer cells, BCSCs were sorted from MCF7/C6 cells and additionally sorted by HER2 expression. The proteomics of HER2+/CD44+/CD24−/low versus HER2−/CD44+/CD24−/low BCSCs was conducted with two-dimensional differential gel electrophoresis (2-D DIGE) and high-performance liquid chromatography tandem mass spectrometry (HPLC/MS-MS) (Duru et al. 2012). Proteins involved in tumor metastasis, apoptosis, mitochondrial function, and DNA repair were enhanced and the HER2–STAT3 network was identified in the HER2+ BCSCs (Duru et al. 2012). Recently, Yun et al. (2016) reported that a radioresistant H460 (RR-H460) cell line derived from radiosensitive H460 lung cancer cells after chronic FR expressed stem cell markers, CD44, Nanog, Oct4, and Sox2 and enhanced aggressive growth and radioresistance with a short list of new genes detected. Depletion of these genes radiosensitized RR-H460 cells. Additional information by mass spectrometry-based proteomic techniques for profiling radioresistant biomarkers has been recently summarized (Chang et al. 2015) including the proteomics of CSCs (Skvortsov et al. 2014). These studies will continue to provide new information required to accelerate the identification of effective targets for radiosensitization of CSCs.
2 CSCs in the Irradiated Tumor Microenvironment
2.1 Warburg Effect and Its Revisions
Tumor cells require increased adenosine triphosphate (ATP) to support their enhanced anabolism and proliferation (Robertson-Tessi et al. 2015; LeBleu et al. 2014; De Luca et al. 2015; Favre et al. 2010; Sotgia et al. 2012). Two bioenergetics pathways are utilized in mammalian cells to provide cellular fuel demands dependent on oxygen status. Under oxygenated conditions cells can metabolize one molecule of glucose into approximately 34 molecules of ATP via oxidative phosphorylation (OXPHOS) in the mitochondria, producing the major cellular fuel for energy consumption. In contrast under hypoxic conditions, cells metabolize one molecule of glucose into two molecules of lactate and two molecules of ATP. In 1956, Otto Warburg discovered that cancer cells tend to convert glucose into lactate to produce energy rather than utilizing OXPHOS even under aerobic conditions. Warburg’s seminal finding has been observed in many tumors that showed mutations in mtDNA and mitochondrial alterations. However, in contrast to Warburg’s original hypothesis, many tumor cells showed intact mitochondria function with inducible MnSOD activation and ATP generation (Guo et al. 2003; Gao et al. 2009; Wallace 2012). In fact, fast growing cells showed enhanced mitochondrial metabolism to meet the challenges of macromolecular synthesis. Recently, CDK1 controlled mitochondrial ATP generation which is linked to normal cell cycle progression (Wang et al. 2014), is also involved in mitochondrial bioenergetics required for cellular fuel demands for DNA damage repair and cell survival after radiation (Alexandrou and Li 2014; Lu et al. 2015; Qin et al. 2015), indicating that the mitochondria in cancer cells, especially in the fast-proliferative tumor cells, can be revivid in generating additional cellular fuels for the increased demands of cellular energy consumption, which is to be further investigated.
2.2 IR Induced Cellular Energy Reprogramming
Growing evidence demonstrate that mitochondria are functional in tumor cells and responsible for metastasis and therapy-resistance (Duru et al. 2012, 2014; Lu et al. 2015; Candas et al. 2014; Obre and Rossignol 2015; Chae et al. 2012; Kang et al. 2014). Although mitochondrial dysfunction is linked with radioresistance of some tumor cells (Lynam-Lennon et al. 2014), mitochondrial bioenergetics is shown to be required to boost cellular fuel production for repairing DNA damage and cell survival, and mitochondrial MKP1 is a target for therapy-resistant HER2-positive breast cancer cells (Lu et al. 2015; Candas et al. 2014). A dynamic feature in mitochondrial bioenergetics which has been observed suggests that CDK1 can boost mitochondrial ATP for cell cycle progression and that cancer cells can quickly adjust cellular energy metabolic pathways to enhance their survival under genotoxic stress conditions such as IR (Fig. 2) (Alexandrou and Li 2014; Lu et al. 2015; Qin et al. 2015; Candas and Li 2014). Heat-shock-protein-90 (HSP90) is involved in proper protein folding in mitochondria that is required for cellular bioenergetics in tumor cells (Chae et al. 2012). mTOR, a critical regulator in cell proliferation, is shown to enhance OXPHOS with reduced glycolysis for tumor cells to survive IR. Thus the apparent “quiet” mitochondria in tumor cells can function as a backup to boost cellular fuel supply required for crisis conditions such as IR. Such a pathway for mitochondrial bioenergetics demonstrates the flexibility of energy metabolism pathways in cancer cells (Lu et al. 2015). Additional findings suggest that mitochondria in tumor cells remain functional and play a key role in tumor cell proliferation and metastasis (Zhang et al. 2013; Park et al. 2016). The dynamic programming of energy metabolism in tumor cells especially in CSCs and, iIR-associated cellular energy metabolism as it relates to tumor cell survival undergoing radiotherapy are currently being investigated. These observations are further supported by findings that reprogramming the mitochondrial trafficking can help to fuel tumor cell invasion (LeBleu et al. 2014; Caino et al. 2015), and that mitochondrial respiration is activated in irradiated tumor cells for survival (Lu et al. 2015). Most importantly, mitochondrial energy metabolism has been recently linked with the aggressive phenotype of triple-negative breast cancer (TNBC) (Park et al. 2016).
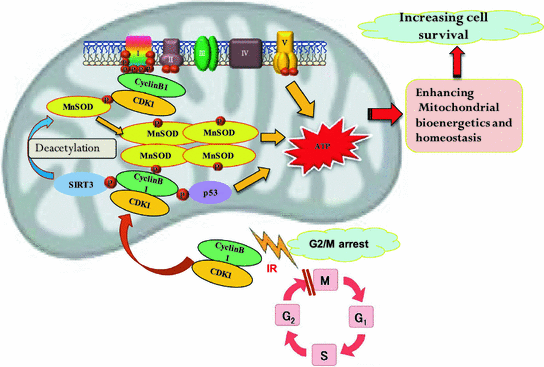
Fig. 2
Proposed mechanism causing mitochondrial bioenergetics for cell survival after radiation
2.3 Metabolic Plasticity of NSCS and CSCs
Accumulating data have recently provided a clearer understanding of energy metabolism in normal stem cells (NSCs). When NSCs divide into two daughter cells, older mitochondria are allocated into one cell and younger ones are apportioned to another daughter cell so as to maintain stemness in one cell and the other with lineage-specific differentiation (Katajisto et al. 2015; Rossi et al. 2007). The amount of mtDNA and mitochondrial biogenesis are gradually enhanced with the increasing cellular energy demands of the cell during lineage differentiation (Cho et al. 2006; Chung et al. 2010). During this process, the typical spherical and cristae-poor mitochondria of undifferentiated stem cells are transformed into tubular and cristae-rich mitochondria which is required to provide adequate ATP for energy metabolism (Chung et al. 2010). It has been noted that gene production for mitochondrial respiration and ROS metabolism are activated and genes related to glycolysis are down-regulated (Chung et al. 2010; Zhang et al. 2013; Urao and Ushio-Fukai 2013; Armstrong et al. 2010; Yanes et al. 2010), indicating a reprogramming from glycolysis to mitochondrial OXPHOS in NSC differentiation (Panopoulos et al. 2012; Lunt and Vander Heiden 2011; Wanet et al. 2014).
Fatty acid oxidation (FAO) produces one molecule of AcCoA in each cycle and two molecule of AcCoA in the final cycle. The AcCoA-induced oxaloacetate produce the citrate for the generation of NADPH (Carracedo et al. 2013). Thus, FAO participates in maintaining sufficient levels of ATP and NADPH in metabolic stress (Pike et al. 2011; Jeon et al. 2012). Unsaturated fatty acids impair the NSC lineage differentiation (Yanes et al. 2010), and other amino acids and TCA-associated metabolism has been linked with NSC self-renewal and differentiation (Lu et al. 2012).
It is far from clear if energy metabolism in CSCs follow the same metabolic adjustments during their self-renewal and/or potential cancer cell differentiation. The question is why cancer cells as well as CSCs can survive the hostile environment such as low nutrition and pH, and even worse situations such genotoxic IR and/or chemotherapy. CSCs can be induced to differentiate into lingual cell population under normal or stress conditions (Vermeulen et al. 2008), and surprisingly, it has been shown that non-cancer stem cells can also be induced back to the stem like feature by radiation with altered cellular bioenergetics (Vlashi and Pajonk 2015). GSCs are found to be less glycolytic and consume less glucose and produce less lactate while maintaining higher ATP levels than their differentiated progeny. However, a higher mitochondrial reserve capacity is detected in GSCs that show radioresistance (Vlashi et al. 2011). Metabolic differences are also identified in BCSCs and differentiated progeny (Vlashi et al. 2014). The metabolic profile which distinguishes the undifferentiated state from the differentiated state of stem cells features a dynamic mitochondrial morphology and a shift from glycolysis to mitochondrial OXPHOS (Westermann 2010; Ferree and Shirihai 2012; Simsek et al. 2010; Suda et al. 2011; Takubo et al. 2013). IR activates mitochondrial OXHPOS (Lu et al. 2015; Candas et al. 2013, 2014), suggesting that mitochondrial bioenergetics can be activated in tumor cells when exposed to genotoxic conditions such as a therapeutic. Thus, mitochondria in tumor cells are functional and can be activated by IR in radioresistant cancer cells and CSCs (Lu et al. 2015; Candas et al. 2014; Candas and Li 2014). A specific population of BCSCs has been identified from surviving breast cancer cells treated by FR (Li et al. 2001; Wang et al. 2005) indicating that BCSCs even from HER2-negative or triple-negative breast cancer (TNBC) cells express not only BCSCs biomarkers (Phillips et al. 2006; Al-Hajj et al. 2004; Reya et al. 2001; Farnie et al. 2007; Cao et al. 2009) but also HER2 (Duru et al. 2012, 2014; Cao et al. 2009). Further analysis revealed that the HER2+ BCSCs are more abundant in the recurrent/metastatic lesions compared to the primary tumors (Duru et al. 2012, 2014) (Fig. 3). All of these results suggest a similar NSC reprogramming of energy metabolism from glycolysis to mitochondrial respiration when CSCs are undergoing differentiation and/or fast proliferation. However, the precise dynamics of cellular bioenergetics in CSCs, especially in vivo under anti-cancer therapy conditions, needs to be further investigated.
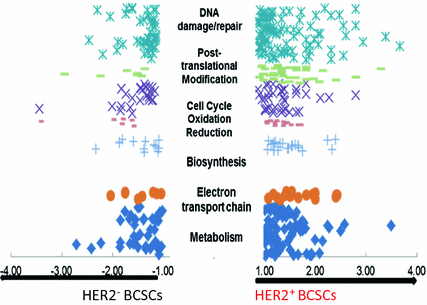
Fig. 3
Proteomics data indiction enhanced proteins in mitochondrial bioenergetics, cell cycle and DNA repair in BCSCs expressing HER2 using HER2negative BCSCs as control
2.4 Hypoxia as a Biomarker for Radioresistant Cancer Stem Cells
It has long been known that there are hypoxic areas within a solid tumor and that radiation induces re-oxygenation (another R in tumor radiobiology) (Brown and Giaccia 1994). Accumulating data demonstrate a wide range of effects by hypoxia which is associated with chemotherapy and radiation resistance, epithelial–mesenchymal transition (EMT), and tumor metastasis (Erler et al. 2006; Xing et al. 2011; Nantajit et al. 2015). The complexity in tumor metabolism is also linked with the hypoxic and nonhypoxic regions within the tumor as well as the surrounding stroma (Dang 2010). Higher levels of HIF-1α in tumors are associated with a poorer prognosis and up-regulation of markers of EMT due to HIF-1α actions. CSCs are believed to reside in a specific microenvironmental niche in the tumor which is required to maintain the CSC characteristics as well as its potential for self-renewal, metastasis and chemo-radioresistance (Peitzsch et al. 2014). Hypoxia-resistant metabolism has been shown to contribute to the aggressive phenotype in ovarian cancer stem cells (Liao et al. 2014), and GSC (Heddleston et al. 2009). Moreover, hypoxia has been shown to induce non stem cells to acquire GSC characteristics with increased tumorigenesis (Zanotto-Filho et al. 2012). The potential functions of HIF-1α and ROS in cancer and pluripotent stem cells have been summarized (Saito et al. 2015). However, there is limited clinical evidence to date to demonstrate that targeting hypoxic regions during conventional therapy is effective. Gene expression signatures of BCSCs and progenitor cells do not exhibit features of Warburg metabolism (Gordon et al. 2015) and oxygen levels do not determine BCSCs radiation survival (Lagadec et al. 2012). Nevertheless, improved image guided individualized hypoxia targeted therapy directed against appropriate molecular targets may significantly enhance the RT efficacy and eliminate CSCs in the hypoxic areas (Peitzsch et al. 2014; Sheehan et al. 2010). It is felt that if more advanced hypoxia-imaging technologies can be developed to visualize the dynamic events of re-oxygenation and/or de-oxygenation during RT, that this would allow one to monitor hypoxic regions together with CSC-repopulation within the tumor under treatment and thus may enhance the accuracy and ability to target resistant tumor cells in the hypoxic regions.
2.5 Abscopal Effect and IR-Induced Immunoregulation
The abscopal effect, described by Nobler in 1969 (Nobler 1969), refers to the potential inhibition of metastatic lesions at a distance from the tumor site being irradiated. The abscopal effect has recently been further highlighted by a series of studies reported by the Formenti’s group demonstrating the benefits of radiotherapy with activation of the immune system by Ipilimumab which inhibits CTLA-4, and also RT combined with granulocyte-macrophage colony-stimulating factor in patients with metastatic solid tumors (Postow et al. 2012; Golden et al. 2013). In the light of the recent growing interest in cancer immunotherapy, there has been increasing interests in the role of the tumor microenvironment on tumor progression and metastasis (Shih et al. 2010; Stefanovic et al. 2014). Although individual immune checkpoint inhibitors have shown clinical benefit, combining these inhibitors with radiation therapy has further enhanced the anti-cancer efficacy. The potential benefits of radiotherapy combined with immunotherapy has been observed (Demaria et al. 2006). In a Phase I clinical trial of 22 patients with metastatic melanoma, radiation combined with inhibition of the programmed cell death protein 1 (PD1) ligand 1 (PDL1)-mediated and CTLA4-mediated immune checkpoints enhanced the tumor responsiveness to immunotherapy. Radiotherapy with double checkpoint blockage of CTLA-4 and PDL-1 enhances T-cell mediated anti-tumor responses outside of the irradiated area (Twyman-Saint Victor et al. 2015). Radiation enhances the diversity of the T-cell receptor (TCR) repertoire of intratumoral T cells (Twyman-Saint Victor et al. 2015). The tumor environment after local irradiation has been suggested to play a key role in tumor immune response after radiation (Golden et al. 2015).
2.6 Tumor Radio-Immunogenicity
The term “tumor radio-immunogenicity” is used here to refer to the observation that specific tumor antigens or epitopes are enhanced or induced after radiotherapy which can then alter the local or systematic tumor immune response. It has been shown that tumors are able to generate an immunosuppressive microenvironment protecting them from host immune surveillance (Schreiber et al. 2011). The selective modulation of Treg in irradiated tumors has been observed which illustrate the immunoregulation occurring in the irradiated tumor microenvironment (Schaue et al. 2008). Radiation induced tumor immune responses are further demonstrated by the fact that irradiated tumors release pro-inflammatory factors such as HSP70 and CXCR6, both of which attract NK cells into the irradiated local tumor environment (Foulds et al. 2013). Derer et al. (2015) recently proposed that increasing the immunogenicity of cancer cells after radiation therapy should be considered as a strategy for systemic cancer immunotherapy and have demonstrated that IR induced tumor phenotypic and tumor microenvironment changes which rendered the cancer cell to be more immunogenic. One of the key immunosuppressive functions of tumor cells has been linked to the expression of CD47, which provides a survival advantage to cancer cells, in particular in CSCs as reported in leukemia, lymphoma, and bladder carcinoma (Chao et al. 2011; Majeti et al. 2009). CD47 is a widely expressed transmembrane protein with multiple functions (Willingham et al. 2012), one of which is to provide a “don’t eat me” signal to phagocytic leukocytes. Phagocytosis by macrophages depends on macrophage recognition of pro-phagocytic (eat me) and anti-phagocytic (don’t eat me) signals expressed on target cells. CD47 on target cells interacts with the ligand signal regulatory protein α (SIRPα) on macrophages (Willingham et al. 2012; Brown and Frazier 2001) resulting in the phosphorylation of the cytoplasmic tail of SIRPα to initiate a signaling cascade that inhibits phagocytosis (Willingham et al. 2012; Zhao et al. 2011). As expected, anti-CD47 monoclonal antibodies have been shown to enhance macrophage-mediated phagocytosis of cells in an array of cancers including bladder cancer, leukemia, and lymphoma (Willingham et al. 2012; Chan et al. 2009; Majeti et al. 2009). In addition, IR-induced immunogenic cell death (ICD) is believed to make the cells ‘visible’ to the immune system for phagocytosis and in initiating other antitumor responses (Galluzzi et al. 2012). However, recent data have revealed another side to this. In addition to IR induced abscopal effects, IR also induces CD47 expression to make the tumor cell “invisible” for phagocytosis enabling them to escape immune surveillance and thus may severely compromise the abscopal effect. Data from the author’s lab support that NF-κB can co-regulate CD47 and HER2. Co-expression of CD47 and HER2 is a key feature of IR-induced adaptive resistance (Fig. 4). Radiation combined with immunotherapy using anti-CD47 and anti-HER2 showed the most synergy in eliminating clonogenic cancer cells, and local tumor radiation was enhanced by application of anti-CD47 antibody (unpublished data). These results demonstrate the complexity of tumor acquired immunotolerance due to activation of the NF-κB-HER2-CD47 pathway which may be a dominant feature in irradiated CSCs. Thus, a dual inhibition of CD47 and HER2 may enhance the abscopal effect. In addition to the above mentioned HSP70, CXCR6, CD47 and HER2, the specific antigens or epitopes that are enhanced or induced in tumor cells by radiation in CSCs are potential therapeutic targets and thus need to be further explored.
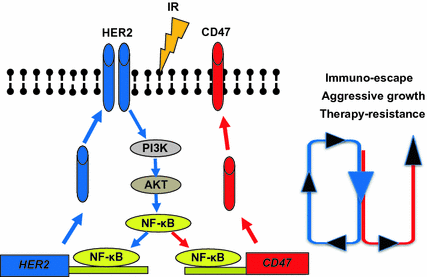
Fig. 4
Proposed co-induction of CD47 and HER2 via NF-κb regulation due to NK-κB binding to both promoters of CD47 and HER2. HER2 is shown to activate NF-κB and radiation accelerates this feed forward loop to further enhance the expression of both. Thus CD47 eqiups the breast cancer cells with HER2-mediated intrinsic pro-survival networks, contributes to the complexity of turn tumor resistance. Radiation combined with immune blocking of CD47 and HER2 may serve as an effective approach to control hard-to treat breast cancer
3 Potential Targets for Radiosensitization of CSCs
3.1 DNA Repair
It has long been proposed that a balance between the degree of DNA damage and activation of pro-survival signaling pathways determines the fate of an irradiated cell. Gene microarray data showed that DNA repair genes are enhanced in the radioresistant fraction of breast cancer cells that survive fractionated radiotherapy (Li et al. 2001). Bao et al. (2006) demonstrate the enhanced DNA repair capacity in GSCs. They also found that the mechanism of radioresistance involves the cell-cycle regulating proteins Chk1/Chk2. Yin et al. showed that ataxia telangectasia mutated (ATM) signaling contributes to radioresistance in CSCs (Frosina 2009). D’Andrea et al. (2011) demonstrated that radioresistance in mesenchymal CSCs is likely due to N-methyltransferase (NNMT) overexpression that is associated with DNA repair mechanisms. Therefore developing small molecules that can specifically bind to CSCs and inhibit the enhanced DNA repair capacity in CSCs will enhance the potential of eliminating CSCs by radiotherapy.
3.2 ROS
Altered ROS metabolism and hypoxia are major features of CSCs. Similarly to NSCs, some CSCs in tumors show lowered ROS levels with enhanced ROS defenses compared to their non-tumorigenic cells (Diehn et al. 2009). Recent studies demonstrate that nuclear factor-erythroid 2-related factor 2 (NRF2), a key regulator of cellular antioxidants and ROS level, is actively involved in maintaining the stemness feature of CSCs (Ryoo et al. 2016). NRF2 regulated ROS level is linked with the growth and resistance of CSCs (Ryoo et al. 2016; Ding et al. 2015). CD44 mediated redox regulation is also suggested in CD44 variant isoforms (Nagano et al. 2013). Blazek et al. showed that Daoy medulloblastoma cells that express CSC features with a low level of ROS can be enhanced under hypoxic condition (Blazek et al. 2007). In agreement with this, Kim et al. (2012) indicated that tumor cells expressing increased CD13 have reduced ROS levels which enhanced the growth of liver CSCs via an EMT-like phenomenon. These results show that ROS levels and redox imbalance are tightly associated with the proliferation status of CSCs which may be altered by IR. Thus the dynamic features of ROS in treated tumor environments need to be further elucidated which may identify approaches to radiosensitization of CSCs.
3.3 MicroRNA
MicroRNA (miRNA; miR) is a small non-coding RNA molecule (containing about 22 nucleotides) involved in gene expression regulation via RNA silencing and post-transcriptional regulation. As epigenetic gene regulators, miRNAs are associated with tumor initiation and progression. An increasing number of reports suggests that miRNAs are promising therapeutic targets of CSCs via epigenetic modification. The function of miRNA in regulation of CSCs is felt to involve a wide array of biological processes involved in CSCs and tumorigenesis (DeSano and Xu 2009; Leal and Lleonart 2013). However, the exact mechanisms underlying miR-regulated CSC biology and therapy-resistance remain to be elucidated. The function of miRNA in NSCs is also linked to CSCs. In addition, miR-504 down-regulates nuclear respiratory factor 1 leading to radioresistance of nasopharyngeal carcinoma (Zhao et al. 2015). It has been shown that the miRNA-153/Nrf-2/GPx1 pathway is involved in the radioresistance and stemness of GSCs (Yang et al. 2015); and knockdown of miR-210 suppresses the hypoxic GSCs and radioresistance (Yang et al. 2014). A nanoparticle conjugated with miR-200c radiosensitized three gastric cancer cell lines and suppressed CD44 and CSCs (Cui et al. 2014).
3.4 IFN-β
Happold et al. reported that GBM cell lines and GSCs (glioma-initiating cells, GICs) express receptors for the immune modulatory cytokine IFN-β. IFN-β treatment remarkably reduced the tumor sphere formation by GSCs (Happold et al. 2014). IFN-β also sensitized GSCs to temozolomide and irradiation. Gene expression profiling showed that IFN-β-associated proapoptotic gene cluster, but not stemness-associated genes were upregulated. Additional analyses revealed the death sensitization mediated by IFN-β is unrelated to chemotherapy or irradiation, indicating that IFN-β is a potential specific GSC target that may be considered in RT of GBM.
3.5 NF-κB and HER2 Crosstalk Pathway
NF-κB activation plays a crucial role in tumor aggressiveness and resistance to anti-cancer therapy (Orlowski and Baldwin 2002; Karin et al. 2002; Bivona et al. 2011; Li et al. 1997). Overexpression of HER2 not only increases cell proliferation and survival (Kurokawa and Arteaga 2001), but also causes NF-κB activation via PI3 K/Akt pathway, which can be inhibited by the tumor suppressor phosphatase PTEN (Pianetti et al. 2001). Radiation induced NF-κB activation can be mediated via nuclear DNA damage through activation of the DNA damage sensor protein ATM (Ataxia Telangiectasia Mutated) (Curry et al. 1999; Locke et al. 2002), and blocking NF-κB inhibits the cell malignant phenotype and radiosensitizes tumor cells (Li and Karin 1998; Brach et al. 1991; Luo et al. 2005; Fan et al. 2007; Ahmed et al. 2006; Braunstein et al. 2008). Rinkenbaugh and Baldwin (2016) recently summarized the NF-κB signaling network in CSCs. Breast cancer cells expressing HER2 were shown to enhance IR-induced NF-κB activation (Guo et al. 2004) and NF-κB in return enhances HER2 gene transcription by activation of ErbB2 promoter (Cao et al. 2009), indicating a positive feedback loop between NF-κB and HER2 (Ahmed et al. 2006; Ahmed and Li 2008). Importantly, HER2-expressing BCSCs (HER2+ BCSCs) are identified in even HER2-negative breast cancer cells that can survive FR (Duru et al. 2012). Recently, persistent activation of NF-κB in BRCA1-deficient mammary progenitors is linked with aggressive phenotype (Sau et al. 2016). In HER2-driven breast cancer mouse models NF-κB pathways contribute to stemness and tumor formation. The canonical NF-κB pathway is required for formation of luminal mammary neoplasia and is activated in the mammary progenitor population (Pratt et al. 2009). Inhibition of NF-κB in a HER2 breast cancer mouse model indicate alterations of gene expression profiles associated with stem cells with NF-κB-dependent changes in the specific stem cell factors Nanog and Sox2 (Liu et al. 2010). Knock-in of a kinase dead IKK led to decreased self-renewal and senescence under mammary stem cell culture conditions (Cao et al. 2007). Thus, it is highly possible that NF-κB and HER2 are mutually activated in CSCs under RT.
3.6 HER2
The HER2 proto-oncogene is located in the long arm of human chromosome 17 and encodes a 185 kD transmembrane glycoprotein in various tissues of epithelial, mesenchymal, and neuronal origin (Soomro et al. 1991; Olayioye 2001). HER2 overexpression is associated with aggressive tumor growth, resistance to treatment, metastasis and a high risk of local relapse and recurrence resulting in poor prognosis (Slamon et al. 1987; Haffty et al. 1996; Holbro et al. 2003). HER2 is valuable both as a prognostic marker and as a predictive factor for therapy response (Haffty et al. 1996; Hicks et al. 2005). The stemness and progenitor cells are increased in normal mammary epithelial cells when HER2 expression is enhanced. The tumorgenicity is also increased with expression of HER2 and ALDH1 (Diehn et al. 2009). These data are significant given that ALDH1 has been suggested as a CSC marker, including in breast cancer (Diehn et al. 2009; Ginestier et al. 2007). Therefore targeting HER2 in breast cancer RT is a promising approach to eliminate HER2-expressing BCSCs, especially for late phase metastatic lesions which are usually multiple tumors and resistant to chemo and radiotherapy.
3.7 MUC13
MUC13 is a transmembrane mucin glycoprotein, is enhanced in many cancers and can activate NF-κB activation. Elevated MUC13 and NF-κB is correlated with colorectal cancer progression and metastases (Sheng et al. 2016). Silencing MUC13 abolished chemotherapy-induced enrichment of CD133+ CD44+ cancer stem cells, slowed xenograft growth in mice, and synergized with 5-fluourouracil to induce tumor regression. Therefore, these data indicate that combining chemotherapy and MUC13 antagonism could improve the treatment of metastatic cancers.
3.8 Wnt/β-Catenin
The Wnt/beta-catenin signaling pathway is associated with the self-renewal of CSC (Reya et al. 2001; Holland et al. 2013). Over-expression of activated β-catenin expands the pool of stem cells (Reya et al. 2003) since it is believed that activation of Wnt leads to the accumulation of β-catenin in cytoplasm and then nuclear translocation to regulate the cluster of genes associated with CSC self-renewal. In a study of myelogenous leukemia, β-catenin was found to accumulate in the nuclei of granulocyte–macrophage progenitors enhancing self-renewal (Jamieson et al. 2004). Thus, dysregulation of Wnt/beta-catenin signaling pathway may be one of approaches worthy of further testing in combination with RT since the self-renewal CSC capacity is enhanced in radioresistant tumors.
3.9 CXCR4
The chemokine C-X-C motif receptor 4 (CXCR4) is found to be a prognostic marker in various types of cancers and is linked with tumor stemness. Interaction of CXCR4 with its ligand, the chemokine C-X-C motif ligand 12 (CXCL12) is believed to function in regulating CSCs and tumor microenvironment. Blocking the CXCR4/CXCL12 axis in CSCs has been evaluated for radiosensitization of CSCs and tumor microenvironment in response to irradiation (Trautmann et al. 2014).
3.10 14-3-3ζ
14-3-3ζ is related to many cancer survival cellular processes. Lee et al. showed that inhibition of 14-3-3ζ reduces the radioresistance of CSCs in HCC (Lee et al. 2014) with reduced capacity of tumor sphere formation and enhanced apoptosis in liver CSCs, indicating that 14-3-3ζ is a candidate CSC target to radiosensitive liver cancer.
3.11 Integrin α6
Integrin α6 which is linked with tumor aggressiveness is found to co-express with GSC markers and enriched in the GSC population (Lathia et al. 2010). Blocking Integrin α6 in GSCs inhibits self-renewal, proliferation, and tumorigenesis of GSCs. These results provide evidence that Integrin α6 can serve not only as an enrichment marker of GSCs but also as a promising radiosensitizer for GSCs.
3.12 YY1
YY1 is a zinc finger transcription factor involved in the regulation of cell growth, development, and differentiation (Seto et al. 1991; Ye et al. 1996). It has also been studied as a potential therapeutic target for anti-cancer therapy (de Nigris et al. 2010; He et al. 2011). YY1 expression is linked with the stemness genes including SOX2, OCT4, BMI1, and NANOG. Proteomics data indicate that co-expression of YY1 and SOX2 as well as SOX2 and OCT4 is regulated by NF-κB pathways (Kaufhold et al. 2016). Therefore, it is possible that dual inhibition of YY1 and NF-κB could be an effective approach to sensitize CSCs to RT.
3.13 Notch
The Notch signaling pathway regulates a wide range of cellular functions in organ development and tissue renewal, and is also highlighted in cancer development due to abnormal Notch functions (Farnie and Clarke 2007). Notch is shown to be able to promote self-renewal and proliferation of mammary stem cells (Dontu et al. 2004) and breast carcinogenesis (Hambardzumyan et al. 2008). Expression of Notch has been associated with radioresistance of GSCs and a potential target for cancer stem cells (Wang et al. 2010; Shen et al. 2015).
3.14 ALDH1A3
Kurth et al. (2013) described that ALDH activity is involved in the radioresistance of CSCs in HNSCC and its isoform ALDH1A3 expression in head and neck squamous cell carcinoma (HNSCC) is suggested to be responsible for tumor relapse after RT. The CSCs in the radioresistant HNSCC cells (SQ20B/SP/CD44+/ALDH-high) were found to extend G2/M arrest phase after RT. UCN-01, a checkpoint kinase (Chk1) inhibitor, induced the relapse of G2/M arrest and radiosensitization of SQ20B-CSCs. All-trans retinoic acid (ATRA) also resulted in ALDH activity and radiosensitized SQ20B/SP/CD44+/ALDH-high CSCs (Kurth et al. 2013). These results indicate that targeting ALDH together with the inhibition of other cell proliferation factors can radiosensitize CSCs.
4 Conclusion and Perspective
In spite of remarkable advances in the precision of radiation dose delivery, the long-term cancer control by RT remains a challenge. One of the key questions to be addressed in tumor radiobiology is to further elucidate the dynamics of tumor microenvironment causing tumor repopulation and resistance. Although much insight has been gained about cancer stem cells and their resident microenvironment, the complexity of the irradiated tumor microenvironment are just beginning to be understood. The exact molecular mechanisms causing the radioresistant phenotype of CSCs, especially the details of dynamics of cross-talking between CSCs and their stroma cells in the microenvironment, remains to be elucidated. In addition to the well-defined Rs in radiation biology and cancer radiotherapy, such as Repair, Re-oxygenation, Repopulation, accumulating new “Rs” are being defined such as Redox balancing, Reprogramming cellular metabolism, Regulation of immuno-response, etc. With more exciting insights revealed into the biology and radiation response of CSCs, continued efforts are expected to dissect and use the key targets of cancer stem cells and its environment to optimize response to radiotherapy. Further characterization of CSCs interaction with the different components in an irradiated tumor microenvironments will shed new light on the mechanisms underlying tumor adaptive resistance and invent more effective radiosensitizing targets. The most promising topics would be the reprogramming of cellular energy metabolism in cancer stem cells and their resident environment of the “tumor society” which consists of multiple factors involved in radiation response and the fate of irradiated tumor cells. In an era of personalized cancer care, identification of specific tumor biomarkers before, during and after a course of radiotherapy with and without chemotherapy, immunotherapy and other anti-cancer approaches would be important. At this point there are few treatment options that specifically and effectively target CSCs.
Acknowledgements
I regret not being able to cite all the important work done in this area due to space restrictions. I’d like to take this opportunity to thank the invaluable input and discussions from all of my colleagues, collaborators and friends who contributed many novel concepts to my research. I thank the graduate students, postdoctoral fellows, and lab personnel who have been involved in the research projects and performed the major of research work in my lab. The author also acknowledges grant support from the National Institutes of Health RO1 CA133402 and CA152313.
References
Ahmed KM, Li JJ (2008) NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic Biol Med 44:1–13PubMed
Ahmed KM, Dong S, Fan M, Li JJ (2006a) Nuclear factor-kappaB p65 inhibits mitogen-activated protein kinase signaling pathway in radioresistant breast cancer cells. Mol Cancer Res 4:945–955PubMed
Ahmed KM, Cao N, Li JJ (2006b) HER-2 and NF-kappaB as the targets for therapy-resistant breast cancer. Anticancer Res 26:4235–4243PubMedPubMedCentral
Alexandrou AT, Li JJ (2014) Cell cycle regulators guide mitochondrial activity in radiation-induced adaptive response. Antioxid Redox Signal 20:1463–1480PubMedPubMedCentral
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003a) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988PubMedPubMedCentral
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


