Introduction
The spinal cord is a critical site for integration of peripheral sensory information en route to the brain and for regulated dispersal of responses via the motor and autonomic divisions of the peripheral nerve system (PNS). Like the brain, the spinal cord is protected physically by its location within the spinal canal of the vertebral column, where it is ensheathed by the dura mater and pia mater membranes and also by the blood:spinal cord barrier (BSCB) which regulates access of blood borne materials. A brief review of the organization of the spinal cord may help set the stage for consideration of how diabetes impacts spinal cord structure and function.
Each of the 31 segments of the spinal cord (8 cervical, 12 thoracic, 5 lumbar, 5 sacral, and 1 coccygeal) receives peripheral sensory input from a pair (1 on each lateral side) of dorsal (in quadrupeds)/posterior (in bipeds) spinal roots that derive from the dorsal root ganglia that house perikarya of peripheral primary sensory neurons. Motor outflow from the spinal cord is via a pair of ventral (in quadrupeds)/anterior (in bipeds) spinal roots containing axons that project from motor cell bodies located in the ventral horn of the spinal cord. The spinal cord itself is fundamentally divided into gray and white matter ( Fig. 7.1 ). The centrally located gray matter is butterfly shaped with pairs of dorsal and ventral horns connected by the gray commissure arranged around the central canal. Gray matter consists of neuronal cell bodies, their dendrites and axons, associated neuroglia and blood vessels. It is also the place where the central projections of peripheral sensory neurons terminate to form synapses with second order neurons and is divided into 10 zones termed the laminae of Rexed ( Fig. 7.1 , right panel). Gray matter is surrounded by white matter consisting of axons, their ensheathing glial cells (astrocytes, oligodendrocytes, microglia) and blood vessels. White matter is broadly divided into anterior, lateral, and posterior funiculi, which are further subdivided into tracts of axons all traveling in the same direction ( Fig. 7.1 , left panel). The ascending tracts ( Fig. 7.1 , left panel, green/italicized text) consist of axons that send information to the brain, whereas the descending tracts ( Fig. 7.1 , left panel, red text) consist of axons that originate from neuronal perikarya located in the cerebral cortex, midbrain, medulla, and vestibular apparatus. Tracts cross from one side of the spinal cord to the other via the anterior white commissure.
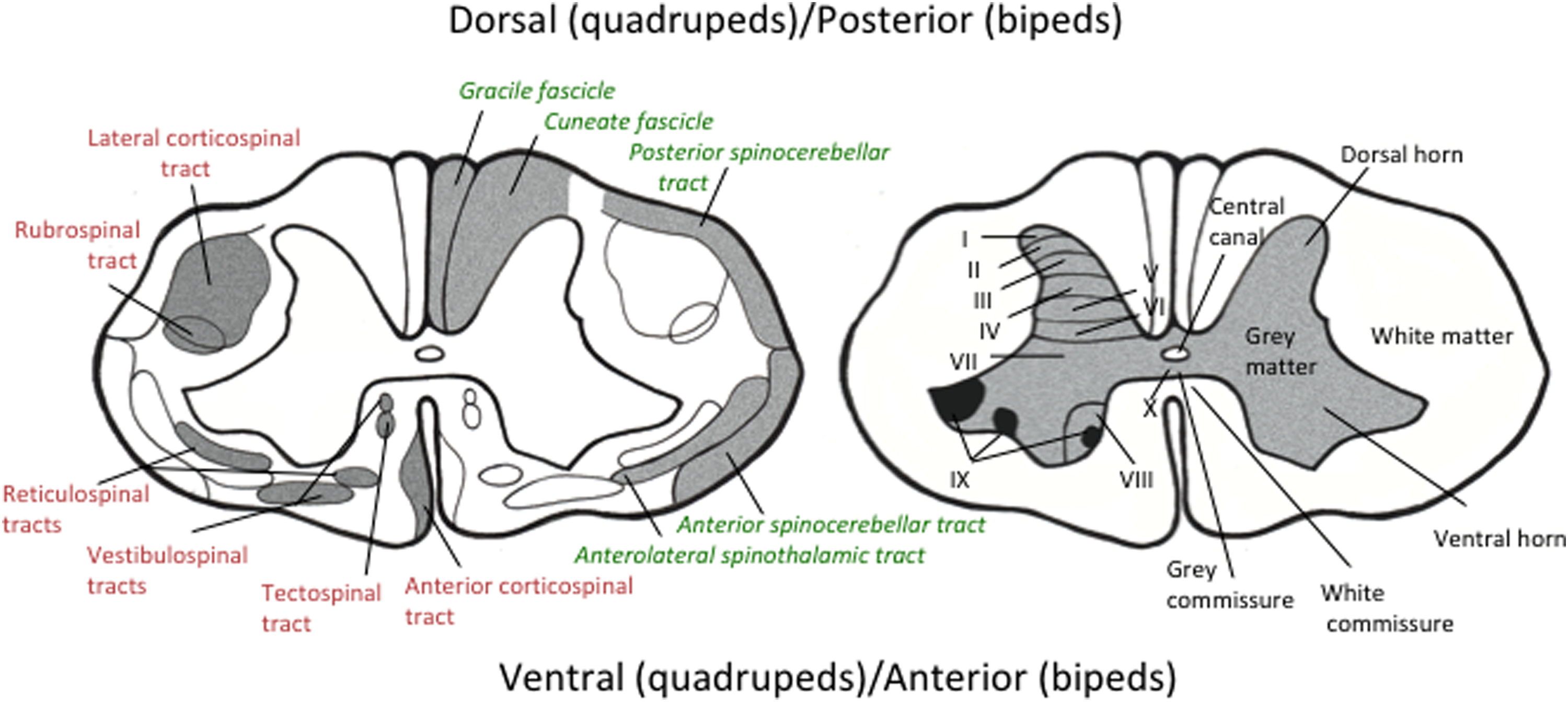
The spinal cord is bathed in cerebrospinal fluid (CSF) of the subarachnoid space that flows down a pressure gradient from the ventricles of the brain and exits via continuities with the endoneurial space of the dorsal/posterior (sensory) and ventral/anterior (motor) roots at the root entry zone. CSF obtained via a spinal tap or intrathecal catheters allows indirect assessment of both local environmental conditions such as glucose levels and also of spinal cord activity via assays of neurotransmitters and metabolites that spill over into the CSF from synapses in the gray matter.
Glucotoxicity in the spinal cord
Existing within the BSCB gives the spinal cord a different exposure to blood borne systemic insults such as the hyperglycemia and dyslipidemia of diabetes compared with peripheral nerve. For example, glucose levels in the CSF are around two-thirds that of peripheral blood and the endoneurial fluid of peripheral nerves. Moreover, although CSF glucose concentrations increase during diabetes , they remain notably lower than those to which peripheral axons and sensory ganglia are exposed via the endoneurial fluid. Consequently, pathogenic mechanisms that rely on tissue exposure to increased absolute glucose concentrations such as nonenzymatic glycation of proteins may not be as toxic to the spinal cord as to peripheral nervous tissue, although cells of the spinal cord will still be exposed to glucose concentrations that are abnormally high in relative terms. Another feature of the anatomy of the spinal cord and associated peripheral nerves is that motor neuron perikarya, located in the ventral horn of the spinal cord, are exposed to lower glucose concentrations during diabetes than are their own axons that project into the periphery. Conversely, the cell bodies of sensory neurons located in the dorsal root ganglia, their peripheral projections and most of their central projections are exposed to more extreme hyperglycemia than the terminals of their central projections that synapse in the spinal dorsal horn and are thus within an environment protected by the BSCB. The sensory-predominant characteristics of some chemotherapy-induced peripheral neuropathies have been attributed to better access of the neurotoxic drugs to sensory ganglia compared to the ventral spinal cord and similar considerations may also apply in diabetes when hyperglycemia or other blood-borne factors are proposed as the initiating toxic insult.
Spinal cord pathology associated with diabetes and neuropathy
Evidence of demyelination, axonal loss, and gliosis has been described in postmortem reports of human spinal cord extending back over a century or more , although caveats concerning tissue source, postmortem delay, handling, and fixation have caused such studies to be viewed with caution. The most commonly reported white matter lesions include axonopathy and myelin thinning/demyelination in the posterior columns, lateral (spinocerebellar) tracts, and ventral columns. In the gray matter, shrinkage, chromatolysis, and loss of anterior horn cells (cell bodies of peripheral motor neurons) with attendant gliosis are most frequently described, with occasional reports of similar damage in the posterior horns. Interestingly, both white and gray matter lesions are more frequent in the lower segments of the cord. This is suggestive of a length-dependent pathogenic mechanism, as it is commonly implicated in diabetic peripheral neuropathy. Functional correlates of spinal pathology may be detected by electrophysiology. For example, somatosensory evoked potential recordings indicate involvement of dorsal column pathways in diabetic subjects with peripheral neuropathy but the alterations are inconsistent with only very minor delays in the spinal conduction of large fiber sensory inputs having been described .
The development of high-quality noninvasive magnetic resonance imaging (MRI) has enabled in vivo assessment of the gross anatomy of the spinal cord of patients with diabetes, thereby precluding concerns about tissue preservation that accompany autopsy studies. Volumetric MRI studies have documented a reduction in cross-sectional spinal cord area in patients with confirmed diabetic neuropathy when compared to age-matched healthy controls and to patients with diabetes and no neuropathy . Spinal cord atrophy was most marked in patients with clinically detectable polyneuropathy compared to those with subclinical disease . This reduction in spinal cord area was seen even at the early stages of neuropathy, suggesting that spinal cord atrophy is an early event in the progression of disease . Unfortunately, current imaging technologies are not able to detail involvement of specific Rexed laminae or white matter tracts or even to identify differential structural involvement of the gray and white matter of the spinal cord and advances in instrumentation will be necessary to provide such detail.
The underlying reason for the reduction in spinal cord cross-sectional area measured by MRI and whether it is a consequence of peripheral nerve pathology or a parallel spinal pathological process is unclear. However, there was a correlation between spinal cord area and sural nerve sensory conduction velocity, as well as with a composite neuropathy score, suggesting that the degree of peripheral and spinal pathology was related and promoting speculation that there may also be shared pathogenic mechanisms. Postmortem findings of microvascular disease in the spinal cord are similar to those seen in peripheral nerves suggesting a shared sensitivity to insults that promote vascular injury. Ischemic hypoxia resulting from microvascular disease has been widely studied as a pathogenic mechanism for diabetic peripheral neuropathy and could also contribute to the pathogenesis of spinal cord lesions . However, the functional significance of such spinal cord pathologies and if they relate to structural and functional alterations in the brain during diabetes remains to be established.
Studies in animal models of diabetes
Many of the genetic, chemical, and dietary animal models of diabetes exhibit multiple indices of peripheral neuropathy and are widely used to investigate pathophysiological mechanisms that may be pertinent to the human condition . With regard to neuropathology, rodent models of diabetes offer the advantage of controlled tissue acquisition and processing for optimal preservation of structures in the spinal cord, which reduces artifacts and allows spinal cord sections cut from resin blocks to be viewed by light and electron microscopy. However, it must also be noted that exposure to diabetes is much shorter in rodents that have a life span of only 1–2 years and are frequently studied after only 1–6 months of diabetes, while the much shorter length of nerves may attenuate pathologies arising from physical length–dependent processes. Both of these features may contribute to the limited number of reports of spinal injury in preclinical studies using diabetic rodents and the relatively mild damage they describe.
Spinal cord pathology
Within white matter, neuroaxonal dystrophy has been described in the dorsal columns, specifically ascending sensory fibers of the gracile fascicle, of spontaneously diabetic rats with a progressive myelinated fiber atrophy and later Wallerian degeneration that presented in a proximal to distal gradient . Autonomic nerves within the cord were also affected, with atrophy of sympathetic postganglionic fibers of the gray ramus despite normal preganglionic fibers in the white ramus . Gray matter pathology is otherwise limited to a report of reduced perikaryal volume of ventral horn motor neurons in the absence of overt degeneration or numerical loss that recapitulated the mild pathology of motor axons in peripheral nerve . Indeed, the absence of overt axonal degeneration and demyelination in the spinal cord of diabetic rodents is generally consistent with the picture in the PNS of these animals, where large fiber axonal atrophy and myelin thinning are the main features seen in nerve trunks following many months of diabetes. The overall pattern of neuropathy in diabetic rodents is one of a slow distal axonal degeneration with spared perikarya .
Spinal cord electrophysiology
Conduction slowing has been reported in both the ascending and descending tracts of diabetic rats, with delayed onset compared to slowing of peripheral nerve conduction . Spinal conduction slowing in type 1 diabetic rats was prevented by spinal infusions of insulin or IGF-1 that was unlikely to modulate systemic hyperglycemia, suggesting a direct neuroprotective effect .
Wide dynamic range neurons, second-order neurons that receive input from a broad range of sensory stimuli and project via the spinothalamic tract to the brain, show abnormal spontaneous activity in diabetic rodents with distinct patterns of dysfunction between different models of diabetes . These neurons also exhibit changes to the shape and organization of their dendritic spines suggesting a structural malorientation that may contribute to abnormal sensory processing . Spinal wind-up, a frequency-dependent increase in the excitability of spinal neurons following repeated electrical stimulation of C fibers, is also enhanced in diabetic mice , further supporting the idea that spinal mediation of sensory processing is altered by diabetes.
Synaptic function in the spinal cord can be assessed using modification of the protocols widely used to measure motor and sensory nerve conduction velocity in rodents, in which stimulation of the sciatic nerve via a needle electrode evokes two waveforms in muscles of the ipsilateral paw; a direct nerve to muscle M-wave and the longer latency trans-spinally mediated H-wave (aka H-reflex: Fig. 7.2 ).
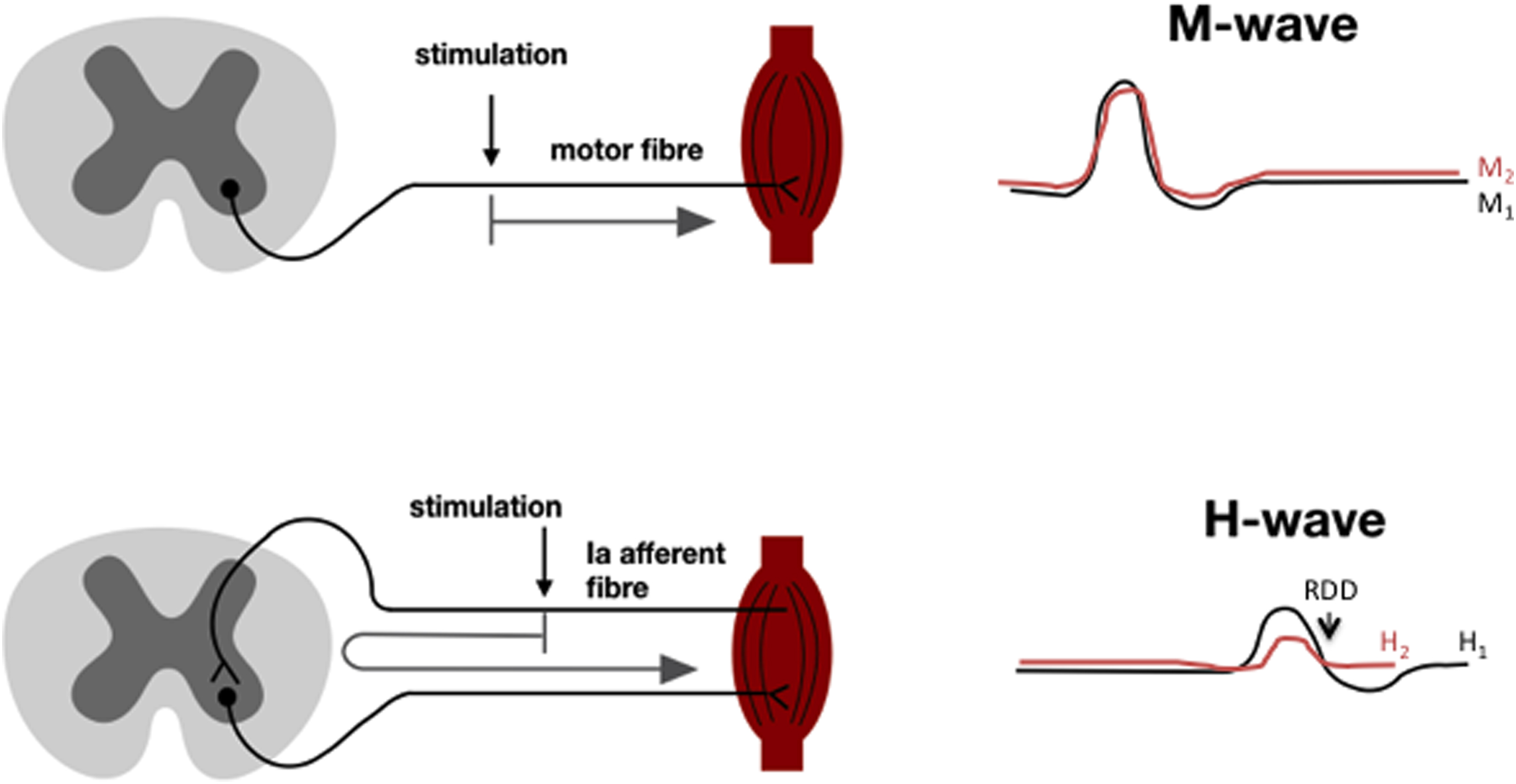
Rather than recording M and H waves evoked at two different stimulation sites and using the peak to peak latency of each wave and distance between stimulation sites to calculate conduction velocity , a train of stimuli from a single location can be used to assess function of the spinal synapse(s) that mediate the H wave. It is well established that stimulus trains of certain frequencies, while having no impact on M wave amplitude, produce a decrease in amplitude of the H wave—phenomenon variously known as rate-dependent depression (RDD or HRDD) paired pulse depression or frequency-dependent depression and that this phenomenon can be lost following physical- or disease-related injury to the spinal cord. Loss of RDD accompanied damage to spinal descending inhibitory systems and interneurons with resulting spasticity that was subsequently linked to disrupted spinal GABAergic inhibition and specifically to failure of the inhibitory functions of the spinal GABA A and ultimately GABA B receptors .
RDD is impaired in the spinal cord of diabetic rodents and humans . In rodents, this has been ascribed to reduced expression and activity of the postsynaptic ion pump potassium chloride cotransporter 2 (KCC2) which disrupts the chloride reversal potential and causes the GABA A receptor, a chloride ion channel, to lose its inhibitory function and develop excitatory function . Excess spinal BDNF has been linked to reduced KCC2 expression . This mechanism is not unique to diabetes and has been advanced as a basic mechanism of spinal sensitization following peripheral nerve injury . Because both RDD and behavioral indices of neuropathic pain in diabetic rodents respond to the same pharmacological interventions and also segregate from neuropathic pain in models of peripherally driven neuropathic pain, it has been suggested that RDD can be used as a biomarker to identify neuropathic pain arising from spinal sensitization in individual diabetic patients . This mechanism is discussed in detail below.
Loss of GABA A receptor inhibitory function may also address another physiological abnormality in the spinal cord diabetic rodents. Studies using spinal microdialysis in conscious unrestrained rats to measure real-time release of neurotransmitters during painful stimuli and subsequent evoked behaviors initially showed that hyperalgesia during the formalin test was unexpectedly accompanied by decreased spinal release of the primary afferent derived excitatory neurotransmitters glutamate and substance P . Reduced substance P release is consistent with the reduced synthesis axonal transport and evoked release of neuropeptides in peripheral sensory nerves of diabetic rodents and their general degenerative phenotype. Prolonged release of prostaglandin E was recorded in the same experimental system, suggesting an extended spinal sensitization response . Moreover, both basal and stimulus evoked release of GABA are also increased in diabetic rats . Excess spinal GABA may therefore drive a spinally mediated hyperalgesia instead of serving its usual inhibitory function. This is supported by the counterintuitive efficacy of GABA antagonists to alleviate behavioral indices of pain in diabetic rats .
Spinal cord cellular pathology
In the absence of overt degenerative pathology in the spinal cord of diabetic rodents, interest has focused on morphological changes to cells and any accompanying shifts in their expression of receptors, cytokines, chemokines, and other molecules. Such changes may indicate early reactive responses to ongoing lesions that precede degeneration or be part of pathogenic cascades that contribute to dysregulation of spinal sensory and motor processing. Indeed, there is a growing appreciation that spinal glia, comprising oligodendrocytes, astrocytes, and microglia, form a network that can orchestrate a coordinated response to insults that includes interaction with the vascular and immune systems and leads to modulation of neuronal synaptic function. This network can also be triggered to driving an inappropriate proinflammatory state in the spinal cord that has been linked to spinally mediated neuropathic pain in many conditions, including diabetes.
Oligodendrocytes
Despite lack of overt demyelination in white matter tracts of diabetic rodents, there is evidence that oligodendrocytes are vulnerable to insults associated with diabetes. Oligodendrocytes show evidence of oxidative damage to myelin lipids and there is reduced expression of myelin basic protein , a protein that interacts with lipids to maintain correct myelin organization. Oligodendrocyte responses to hyperglycemia may also promote spinal modification of sensory processing. For example, oligodendrocytes, like their peripheral nerve counterparts (Schwann cells), express both aldose reductase and cyclooxygenase 2 (COX-2) . Aldose reductase has been widely implicated in the pathogenesis of diabetic neuropathy and other complications due to its activity metabolizing excess glucose during hyperglycemia , while COX-2 produces prostaglandins that evoke inflammatory and some forms of neuropathic pain. Spinal glucose metabolism by aldose reductase during diabetes increases COX-2 expression and activity in oligodendrocytes, which in turn drives prostaglandin release and promotes spinally mediated hyperalgesia . Thus, while the severity of hyperglycemia and duration of diabetes that are achieved in rodent models of diabetes may not be sufficient to cause oligodendrocyte death or demyelination, there are indications of early molecular stresses on oligodendrocytes that may lead to both myelopathy and promote neuropathic pain.
Astrocytes
Astrocytes are distributed throughout the spinal cord, where they associate with neuronal synapses in the gray matter to modulate neurotransmitters and their metabolites. Astrocytic foot processes also regulate the fluid microenvironment of the spinal cord by ensheathing the endothelial cells and pericytes of the blood vessel wall, which together forms the blood:brain barrier (BBB) and BSCB. Electron microscopy studies have suggested that diabetes causes retraction of astrocytic foot processes from the other cells of the BBB . Expression of proteins associated with junctions between adjacent endothelial cells, such as occluding and VE-cadherin , is also reduced in the spinal cord of diabetic rats. At present, the majority of studies of astrocytes in diabetes have focused on the brain, which is not surprising given the emerging association between diabetes-induced macro- and microvascular disease, increased BBB permeability, and cognitive dysfunction . However, data obtained in the brain may not directly extrapolate to the spinal cord given that diabetes-induced BBB permeability shows regional variations even within the brain . Indeed, increased albumin leak through the BBB was reported in the periventricular area of the brain of diabetic mice, but not in the spinal cord of the same animals . Whether this reflects a permanent regional difference or a temporal variation as diabetes-induced lesions evolve is not known. In contrast, Evans blue extravasation into the spinal cord, another measure of BSCB permeability, has been reported to be markedly reduced in diabetic rats—an unexpected finding that was attributed to reduced spinal vascular volume and thus reduced blood flow . Whether this is the result of loss of blood vessels or vasoconstriction has yet to be confirmed by detailed histopathological studies.
A number of studies have measured expression of glial fibrillary acidic protein (GFAP) as an index of astrocyte number and health status. Reduced total spinal GFAP levels and numbers of GFAP expressing astrocytes have been reported in both white and gray matter of streptozotocin-induced type 1 diabetic rats , though whether this reflects reduced expression or cell loss is unclear. Conversely, increased total spinal GFAP protein, GFAP immunostaining intensity, number of GFAP expressing cells, and altered astrocyte morphology have been recorded in the dorsal horn of type 1 and type 2 diabetic mice and rats , along with increased expression of nNOS and iNOS . Such findings are generally interpreted as reflecting initiation of astrogliosis to fill the void of degenerated cells and/or activation of astrocytes as part of a more generalized spinal neuroinflammatory state that, in concert with microglia , contributes to inflammatory-like spinally mediated neuropathic pain .
Microglia
Microglia are the resident macrophages of the CNS and are increasingly recognized as prominent orchestrators of the interactions between neurons, astrocytes, and the immune system in the brain and spinal cord . They are versatile immune cells that possess properties that in the vascular immune system are distributed across multiple specialized cell types, thereby obviating the need for infiltration of hematogenous immune cells into the CNS in response to pathogens, tissue injury, or other lesions. Microglial activation was initially defined as a change in cell morphology with thickening and reduction in the length and complexity of their ramified processes and an increase in cell body size/volume that represents a shift from environmental surveillance (ramified) to injury-evoked response (reactive) . More recently, it has become recognized that the reactive microglial population also exists in a plastic spectrum spanning the two polarities: proinflammatory and phagocytic M1 and anti-inflammatory/neuroprotective M2, and that shifts in population size and ratio can both reflect and drive changing environmental conditions .
Diabetes has been widely reported to activate resident microglia in the spinal cord of rodents, with definition of what constitutes an activated (or more accurately reactive, as ramified microglia are also inherently active) cell varying from the morphological , though the presence of generic signals of cell activity to expression of specific marker proteins that identify cells as being in a specific region of the polarization spectrum. Early reports linked microglial activation by proinflammatory cytokines with kinin B1 receptor expression in the genesis of spinally mediated pain in diabetic rats . Microglia associate with synapses and have the potential to regulate and modify spinal sensory processing. Involvement in the genesis or amplification of pain has been supported by multiple studies showing that disruption or depletion of spinal microglia can prevent or reverse indices of neuropathic pain in diabetic rodents . Reactive microglia release multiple factors that can influence spinal sensory processing, including BDNF (via regulation of KCC2 as discussed above) and proinflammatory cytokines such as IL-1β and TNFα, as recently reviewed .
A proportion of the activated microglia in the spinal cord of diabetic mice may derive from infiltrating blood-derived monocytes , while infiltration of neutrophils into the gray matter of diabetic rats has also been reported and was attributed to increased levels of l -selectin, an adhesion molecule expressed by leukocytes that is involved in their migration from blood vessels into tissue parenchyma . Given the physical restrictions placed on expansion of the spinal cord due to the surrounding bone, it is not clear whether infiltrating cells are replacing other degenerating or departing cells to maintain overall spinal cord volume or precipitating increased intraspinal pressure that itself could have pathological consequences.
Clinical studies of spinally mediated pain in diabetes
Preclinical studies have suggested a variety of mechanisms that may contribute to neuropathic pain arising from peripheral, spinal, and supraspinal sites in models of diabetes that have recently been reviewed in detail elsewhere . The remainder of this chapter will therefore focus on clinical evidence of potential central mechanisms that influence output from nociceptive projection neurons in the dorsal horn of the spinal cord in painful diabetic neuropathy.
The spinal cord and painful diabetic neuropathy
Up to one-third of patients with diabetes, suffer with neuropathic pain that often leads to sleep disturbance, poor quality of life, anxiety/depression, and unemployment . Data suggest that 10-year mortality is higher in patients with severe chronic pain . The only treatment option is pain management, which typically involves a trial and error approach of prescribing antineuropathic pain medications, which themselves have inconsistent therapeutic benefit . Major unmet needs exist for the development of reliable biomarkers that both facilitate individualized treatment of neuropathic pain and inform mechanistic-based drug discovery.
Pain is a complex and subjective physiological and psychological experience and is therefore inherently difficult to study and treat . Why some patients develop pain yet others do not, despite apparently equivalent severity of neuropathy, remains an unanswered conundrum . The individual variability in pain perception poses additional challenges to assessing and treating pain . Sural nerve biopsy studies in patients with diabetes with or without pain show no associations between pain and degeneration and/or regeneration of large or small fibers . However, recent data indicate an association between loss of small fiber peripheral terminals in subjects with both neuropathy and pain when compared to those with equivalent neuropathy but no pain suggesting an association between the degree of small fiber neuropathy and pain . The association of pain with distal fiber loss also supports the long held idea that pain perceived as being in the extremities is generated by metabolic or physical injury in the extremities. However, this is clearly not the whole story. In 1999 Rajbhandari and colleagues described the case of a patient who had undergone a below knee amputation as a teenager because of an arteriovenous malformation. In his 50s, the patient developed typical distal bilateral neuropathic pain in both the amputated and the intact leg, which was closely followed by a diagnosis of type 2 diabetes . This suggests that the neuropathic pain, at least for the amputated limb, originated from somewhere other than the perceived location of the pain.
Peripheral input
Disorders of the PNS can result in hyperexcitability of nociceptors; those results in an increased nociceptive drive to the spinal cord. However, this does not always equate with pain. Indeed, there is often a disconnect between the degree of peripheral nociceptive drive and the severity of perceived pain. It is well known that the severity of perceived pain depends upon many higher CNS factors including level of arousal, anxiety, depression, expectation, and anticipation . There are also neural processes that are mediated by, or are intrinsic to, the spinal cord that may suppress or enhance ascending nociceptive projections.
Primary afferent activity, whether as a result of activation at the peripheral endings or, aberrant spontaneous activity due to peripheral nerve dysfunction, inputs into the spinal cord, with the majority of afferents arbourising and making synaptic contacts in the dorsal horn. Inputs from particular afferent types terminate in distinct Rexed laminae, with nociceptor afferents terminating in the superficial laminae of the dorsal horn . The sensory information resulting from pain-related afferent input is processed in a complex dorsal horn interneuronal network and subsequently further integrated by projection neurons in the superficial and deep laminae of the spinal cord . Thereafter, pain-related signals ascend in distinct spinal cord tracts (see Fig. 7.1 ), most notably via the contralateral spinothalamic tract to the thalamus and brain . There are also direct projections to the brainstem and hypothalamus via the spinoreticular/spinomesencephalic and spinohypothalamic tracts, respectively . Signal amplification or loss of inhibition (disinhibition) could potentially occur at any point along these spinal systems resulting in enhanced nociceptive transmission to the brain.
It is also important to appreciate that the spontaneous and evoked neuropathic pain felt by patients, whether due to pathology/dysfunction/alterations affecting the PNS or indeed CNS, is likely dependent on some form of peripheral input . This does not imply that the stimulus has to be abnormal peripheral activity generated by sensitized nociceptors, but rather that the CNS, even in the case of CNS damage, does not autonomously generate neural activity that is perceived as pain without some peripheral afferent input.
Spinal mechanisms
There are a number of theoretical mechanisms by which peripheral inputs to the spinal cord could be subject to amplification (or reduced suppression) including:
- •
Glial activation within the dorsal horn of the spinal cord
- •
Facilitation due to synaptic plasticity in the nociceptive dorsal horn
- •
Wind-up
- •
A reduction of tonic spinal inhibition (spinal disinhibition)
- •
Alterations in the descending modulation of spinal cord processing and outputs
Glial activation, as discussed above, is currently restricted to an interesting series of preclinical observations that have yet to be validated in humans. There is also no evidence suggesting that synaptic facilitation due to long-term potentiation is of importance in enhancing ascending nociceptive outputs from the dorsal horn in diabetes. Since the induction of long-term potentiation along the pain pathways is classically dependent on high frequency bursts of nociceptive input from the periphery, a situation that is most likely to occur in the context of an acute nerve injury, such a mechanism is also theoretically unlikely to prominent in a chronic neuropathy. We will therefore focus on evidence linking the remaining three potential mechanisms to pain associated with diabetic neuropathy.
Wind-up
Pain wind-up, which is related to the psychophysical phenomenon of pain temporal summation, refers to the facilitation of nociresponsive spinal cord neurons, including spinothalamic projections neurons as a result of from repeated low frequency stimulation of c-fiber nociceptors. It has been proposed as a potential contributing mechanism of allodynia and hyperalgesia . Both wind-up of spinal cord neurons in rodents and temporal summation in human subjects are reduced by NMDA receptor antagonists, although the effect is partial and other nonglutamatergic mechanisms may also contribute.
There have been few reported investigations addressing wind-up in preclinical models of diabetes, with a sole report that repetitive electrical stimuli targeting c-nociceptor afferents in diabetic mice enhanced the nociceptive withdrawal reflex—an intrinsic polysynaptic circuit that mediates removal of a limb from a noxious stimulus . Temporal summation is assessed in humans using repetitive noxious thermal, electrical, or mechanical stimuli. The pain intensity rating typically of the 10th stimulus is compared to that associated with the first stimulus. No differences in mechanical temporal summation have been demonstrated between patients with either nonpainful or painful diabetic neuropathy and healthy controls or between painful and nonpainful diabetic neuropathy suggesting that excessive wind-up is not a major mechanism underlying painful symptoms in humans. However, patients with a shorter duration of painful diabetic neuropathy have significantly more pronounced temporal summation than patients with a longer duration, despite equivalent pain severity, indicating that the spinal mechanisms involved in initiating and maintaining pain may evolve over time as part of what is sometimes termed the chronification of pain .
Impaired tonic spinal inhibition (spinal disinhibition)
A discussed in detail above, there is accumulating preclinical evidence that loss of RDD and indices of neuropathic pain share a common pathogenic mechanism involving spinal KCC2 depletion and disinhibition caused by loss of GABA A receptor inhibitory function and possibly conversion to excitatory functions . As both RDD and behavioral indices of neuropathic pain exhibit common responses to spinally acting analgesics, it has been suggested that RDD status may be a viable biomarker for identifying the dominant generator site in individual patients with painful diabetic neuropathy and for predicting efficacy of therapeutic strategies that alleviate spinal disinhibition.
Loss/impairment of RDD has been demonstrated in spinal cord injured patients, likely attributable to disinhibition of sensory processing . To assess the translational potential of the preclinical findings, we measured the magnitude of H-reflex RDD in patients with type 1 diabetes and painful or painless neuropathy and age-matched healthy controls using a minor modification of the protocol for performing nerve conduction studies . We found that in patients with painful diabetic neuropathy, there was loss of RDD compared to both healthy controls and patients with painless diabetic neuropathy. The impairment of RDD was independent of measures of both large and small fiber neuropathy indicating that is not merely a reflection of severity of neuropathy. Loss of RDD was also independent of glycemic control. Not all subjects with pain had impaired RDD (defined as a H3:H1 ratio >2 × SD of the control group mean) and indeed the degree of RDD overlapped with both healthy controls and patients with painless diabetic neuropathy. These findings support the hypothesis that impaired RDD may serve as clinical biomarker in a subset of patients where pain arises primarily from spinal disinhibition. Furthermore, loss of RDD correlated with pain severity indicating that more severe pain is associated with the presence of loss of RDD and, by inference, spinal disinhibition. Expanding these data from patients with type 1 diabetes, we have recently demonstrated that impaired RDD is also seen in patients with type 2 diabetes and painful neuropathy (unpublished observations). Similar to the findings in type 1 diabetes, not all subjects with type 2 diabetes and neuropathic pain demonstrated impairment of RDD. Approximately 60% of patients with diabetes will develop neuropathy, 30% of those with neuropathy will develop neuropathic pain and, from our exploratory study, 40% of those will show RDD deficits. In contrast, diabetic rodents exhibit much more homogeneous neuropathy, neuropathic pain, and impaired RDD phenotypes . While this may reflect a more complex aetiopathogenesis of painful diabetic neuropathy in humans, it is also plausible that this heterogeneity can be used to enable definition of abnormal values and predict therapeutic response to medications that target spinal inhibition.
It is important to acknowledge that a proportion of patients in our clinical studies of RDD were taking prescribed antineuropathic pain medication and that therapeutic responses were not studied in a systematic manner. Similarly, it is not known if antineuropathic pain medication affects or normalizes RDD either in a general or drug-specific manner. Further larger scale studies that control for the use of antineuropathic pain medication and/or systematically assess therapeutic response are required to validate the use of RDD both as a biomarker of spinal disinhibition and a predictor of neuropathic pain medication efficacy in patients with painful diabetic neuropathy. While the method is noninvasive and potentially widely available, one potential drawback to using RDD as a biomarker is that the H-reflex can be absent or difficult to elicit in a proportion of patients with advanced diabetic neuropathy . Loss of the H-reflex can also be caused by other commonly encountered conditions such as radiculopathies affecting the S1 nerve root . These may limit the use of RDD in patients with severe neuropathy or in those with coexisting lumbosacral root disease.
Descending modulation of spinal cord processing and outputs
It is important to not view processing within the spinal cord in isolation. There are multiple means by which abnormalities in PNS firing/metabolism impact upon the complex interconnected circuits within the dorsal horn. Conversely, there is increasing recognition that top down descending inputs to the spinal cord from the descending pain modulatory system (DPMS) can dynamically modulate ascending sensory information. The DPMS involves a complex brainstem–subcortical–cortical network that projects to the spinal dorsal horn and can have either a facilitatory or inhibitory influence on ascending nociceptive input to the brain . The major outputs of the DPMS arise from the periaqueductal gray situated in the midbrain, which descend via mu-opioid sensitive neurons in the rostral ventral medulla to the spinal cord . A parallel projection also arises from the locus coeruleus . These descending pathways chiefly involve the monoanimergic neurotransmitters noradrenaline and 5-hydroxytryptamine and can have pronociceptive or antinociceptive effects on dorsal horn processing according to the receptor that is activated .
One major descending inhibitory system of interest is the diffuse noxious inhibitory controls (DNICs). DNIC is triggered by a noxious stimulus that results in the activation of descending controls that inhibit WDR projection neurons of the deep spinal cord with receptive fields heterotopic to the conditioning stimulus . This results in a reduction in ascending nociceptive drive. In humans, the efficacy of the DNIC system can be evaluated using a number of psychophysical paradigms, broadly termed conditioned pain modulation (CPM). This involves triggering the DNIC system by delivering a noxious stimulus distant to a control site. For instance, CPM can be easily initiated by immersing one hand in painfully cold ice water while assessing ratings for a painful stimulus on the contralateral side . Impairment of CPM, indicative of altered descending modulation, has been demonstrated in patients with diabetes and painful neuropathy . Furthermore, treatment of painful diabetic neuropathy with duloxetine, a serotonin-norepinephrine reuptake inhibitor thought to augment monoaminergic descending inhibitory pain control, has been shown to be a more effective treatment in patients with reduced CPM . Similarly, treatment with tapentadol, which is also implicated in the modulation of descending pain inhibitory pathways through both activation the µ-opioid receptor activation and inhibition of neuronal norepinephrine reuptake, improved pain in patients with painful diabetic neuropathy and impaired CPM . Interestingly, treatment with duloxetine or tapentadol, in parallel with their beneficial therapeutic effects on pain, also activated CPM .
As well as deficient inhibition, enhanced facilitation could also result in an increase in ascending nociceptive drive from the spinal cord. A recent study that examined subjects with painful diabetic neuropathy by fMRI and quantitative sensory testing found that ventrolateral PAG functional connectivity is altered in patients suffering from painful diabetic neuropathy . Furthermore, the magnitude of ventrolateral PAG connectivity to the rostral anterior cingulate cortex correlated with the degree of evoked pain in response to a tonic thermonoxious stimulus, suggesting that there was a facilitation of nociceptive drive . While the spinal cord was not directly assessed, the importance of the ventrolateral PAG as a key node of the DPMS implies that the findings might relate to enhanced descending facilitation of nociceptive drive form the spinal cord and that this is influenced by higher cortical centers.
It is not known whether pronociceptive alterations in the DPMS precede the development of pain. There are no published prospective studies that address this issue and, furthermore, the current literature is biased toward the testing of patients with painful rather than painless diabetic neuropathy. However, the possibility that the reduced capacity for patients with diabetic neuropathy to activate DNIC might reflect an early phenomenon was suggested in a cross-sectional study in patients with varying duration of neuropathic pain symptoms . Despite the persistence of pain, CPM was shown to become more efficient with a longer duration of diabetic neuropathy. CPM values in patients with a longer duration (>2 years) of diabetic neuropathy were no longer pronociceptive and, indeed, similar to those seen in healthy control participants. This effect that was unrelated to antineuropathic pain medication. This suggests that the influence of DPMS on spinal cord nociceptive processing in painful diabetic neuropathy is dynamic and may return to equilibrium over time. Whether the dynamic alterations in the DPMS that either enhance or suppress inhibition in the dorsal horn of the spinal cord are linked to the mechanisms underlying painful diabetic neuropathy-related spinal inhibitory dysfunction is not known.
Top-down monoaminergic modulation of ascending spinal nociceptive projections is likely to exhibit significant complexity due to multiple parallel descending pathways acting on a multiplicity of targets in the dorsal horn. For example, there is a wide diversity of dorsal horn inhibitory and excitatory interneuron subtypes and the patterns of monoamine receptor subtypes expressed by these interneurons . These interneurons act within a complex network that can modulate projection neurons, other interneurons and primary afferent fibers through GABA A -mediated primary afferent depolarization . There is evidence that monoamines act, in part, by enhancing GABA A transmission in the dorsal horn of the spinal cord. For instance, the application of 5-HT to spinal cord slices has been shown to enhance GABA release . Furthermore, serotonin acting through 5-HT 2 receptors directly modulates GABA A by potentiating GABA evoked inward currents. The antinociceptive effects of serotonin, acting through 5-HT 3 receptors on WDR projection neurons in the dorsal horn are blocked by the GABA A antagonist bicuculline, implying that the monoaminergic inhibitory effects are mediated indirectly via inhibitory interneurons . Similarly, as well as mediating peripheral nociceptive inputs via presynaptic inhibition , noradrenaline also directly excites dorsal horn GABAergic interneurons .
As discussed above, preclinical models of painful diabetic neuropathy indicate that spinal inhibitory dysfunction is driven by GABA, where this normally inhibitory neurotransmitter, acting via ionotropic GABA A receptors, becomes pronociceptive by causing depolarization rather than hyperpolarization of the postsynaptic neuron . If there was significant convergence of GABAergic circuits underlying DPMS and spinal disinhibition, it is conceivable that otherwise normally functioning descending pain inhibitory pathways become facilitatory when acting in the context of spinal disinhibition by enhancing depolarizing GABA transmission. Since both DNIC and spinal disinhibition can be easily measured in humans, using CPM and HRDD, respectively, this could provide a window of opportunity to study the potentially convergent interactions in dorsal horn inhibitory pathways in patients with DPN.
Conclusion
Rather than being merely a passive conduit for motor and sensory tracts, the spinal cord forms a critical sensorimotor processing interface between the brain and the periphery. Despite its protection by physical and physiological barriers, the spinal cord is exposed to, and damaged by, fundamental pathogenic features of diabetes such as impaired insulin signaling and hyperglycemia. Building on traditional pathological and electrophysiological approaches, increasing preclinical and clinical evidence emerging from application of new technologies is strengthening the argument that pathophysiological modifications to the processing of sensory inputs to the spinal cord in diabetic neuropathy contribute to symptoms such as pain.
Future studies in patients with diabetic neuropathy that combine deep phenotyping of neuropathy and neuropathic symptoms along with neurophysiological and psychophysical measurements of spinal cord function are likely to provide further important pathophysiological insights. This approach, supplemented by advancements in noninvasive imaging of the human spinal cord using fMRI, may have direct clinical application by allowing patients with pain mediated by spinal pathophysiological alterations to be treated with therapies that selectively target these mechanisms.
Acknowledgments
Supported in part by award number 1-17-ICTS-062 from the American Diabetes Association (AGM, NAC).
References
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


