The high fever that regularly follows transfusion of some multiparous women and patients who have received many transfusions is often associated with serum leucocyte antibodies; mild febrile reactions, observed after only one of a series of transfusions, commonly are not (Kevy et al. 1962). Such reactions may be mediated by antibodies undetected in standard screening techniques or by passive transfer of cytokines and other pyrogens (see below).
Granulocytes react with anti-HLA-A, -B and -C, as well as with granulocyte-specific antibodies. Monocytes react with the same HLA antibodies, with antibodies against HLA class II antigens and with monocyte-specific antibodies as well. Several studies support the conclusion that HLA antibodies play a role in causing febrile transfusion reactions. Mangano and co-workers (1991) found a reaction rate of 14% after single-donor platelet transfusions that fell to 6.5% when HLA-matched platelets were used, whereas previously Chambers and co-workers (1990) found the rate of unmatched platelets to be almost twice that of matched platelets. In both studies, unmatched pools of platelets resulted in the highest reaction rates. The reduction of febrile reactions with HLA-matched platelets has been confirmed in other studies (Williamson et al. 1994).
Two caveats bear repeating: (1) In patients who have had febrile reactions following transfusions, the presence of serum alloantibodies cannot be excluded with any confidence unless several different assay methods and an extensive panel of cells are used. (2) Antibodies directed against leucocyte antigens, particularly anti-HLA, are common in multiparous women and repeatedly transfused patients. Their presence in patients with fever does imply cause and effect.
Febrile Reactions in Infants
Infants seem incapable of shivering. Perhaps this explains an apparent lack of interest in the infant’s reaction to transfusion. If an infant’s temperature is recorded at intervals after transfusion, it is not unusual to observe rises in temperature up to 38.5°C or thereabouts. Sometimes a temporary refusal to feed and diarrhoea accompany the rise in temperature.
Although an infant does not shiver in the early phase of a febrile reaction, the infant may look pale and its skin may feel cold. The following is an example:
Reactions Following Granulocyte Transfusion
Reactions to transfused granulocytes may be severe and even life-threatening. The majority of these reactions occur in patients who have pre-existing leucoagglutinins, most often HLA antibodies. Reactions have also been associated with isolated granulocyte-specific antibodies. Although fever is the most common sign, additional signs and symptoms include anxiety, sudden hypotension, hypertension, dyspnoea and cyanosis with moderate oxygen desaturation. In a study of 18 patients with chronic granulomatous disease (CGD) and frequent courses of granulocyte transfusions, 14 patients had evidence of leucocyte antibodies measured by lymphocytotoxicity, granulocyte immunofluorescence or granulocyte agglutination assays. Febrile reactions occurred in 11 out of the 14 sensitized patients; seven suffered pulmonary reactions. None of the four patients who lacked antibodies reacted to granulocyte transfusion (Stroncek 1996). A similar case with repeated severe pulmonary compromise was reported when granulocytes were transfused to a patient who was found to have a strongly positive granulocyte crossmatch. That reaction has been categorized as transfusion-related acute lung injury (TRALI) (see below) (Sachs and Bux 2003). Cell labelling studies provide further evidence that these reactions are caused by granulocyte–antibody interaction. The fluorescent label, dihydrorhodamine-123, can distinguish normal granulocytes from those that lack the oxidative burst, such as granulocytes from patients with CGD. Less than 1% of transfused granulocytes could be detected by dihydrorhodamine-labelling in alloimmunized CGD patients who developed reactions during granulocyte transfusion; in contrast, measurable increments were found in patients who sustained no reaction (Heim et al. 2011). Granulocyte circulation, severe febrile and pulmonary reactions may correlate less well with the presence of alloantibodies in patients receiving myeloablative therapy for acute leukaemia and haematopoietic transplant than in those with CGD and aplastic anaemia (Price et al. 2000). Alloimmunized patients who develop fever with granulocyte transfusions are unlikely to derive benefit from this treatment, may become seriously ill as a result of further granulocyte transfusion. Preference should be given to serologically compatible transfusions in the future.
Febrile Reactions Induced by Other Soluble Factors in Stored Blood
Plasma factors have been associated with febrile transfusion reactions for more than 50 years (Dameshek and Neber 1950). As these observations occurred before the era of sterile, interconnected plastic storage bags, the plasma factor(s) may have been bacterial pyrogens, leucoagglutinins or, less likely, cytokines derived from the cellular components. Patients still develop fever after platelet transfusion even if platelet concentrates have been depleted of leucocytes immediately before the transfusion (Mangano et al. 1991; Muylle and Peetermans 1994). Heddle and co-workers (1993) demonstrated that infusion of the plasma supernatant removed from stored platelets is associated with these febrile reactions. Evidence is mounting that many febrile reactions, particularly those related to platelet concentrates, are caused by cytokines, chemokines and possibly other cell-derived soluble factors that accumulate during component storage (Heddle 1999). Viable leucocytes are the prime suspects. Leucocyte removal prior to platelet storage (and prior to cytokine elaboration) appears to reduce the number of reactions. In a randomized study of 1190 transfusions of 129 patients, the frequency of reactions was 13.6% when platelets were leucoreduced prior to storage (Heddle et al. 2002). Severe reactions declined to 1–2%. As might be expected, the frequency of febrile reactions induced by platelet concentrates increases with the length of platelet storage (Muylle et al. 1992; Heddle et al. 1993). Chills, rigors and flushing have also been associated with elevated concentrations of these soluble factors, although these symptoms alone are usually not reported as ‘febrile’ reactions.
The concentration of various cytokines (TNF-α, IL-1α and -β, IL-6) increases during platelet storage and correlates with the number of leucocytes in the concentrate (Muylle et al. 1993; Enright et al. 2003). IL-8 has also been found to increase during storage (Stack and Snyder 1994). Not unexpectedly, stored platelet concentrates prepared by the platelet-rich plasma (PRP) method induce febrile reactions more frequently than do concentrates prepared by the buffy coat method, which contain fewer leucocytes (Flegel et al. 1995; Muylle et al. 1996). The frequency of febrile reactions induced by plasma separated from stored platelet concentrates correlates strongly with the concentration of IL-1 and IL-6 in the plasma (Heddle et al. 1994).
As the leucocytes in a platelet concentrate do not contain detectable amounts of IL-1 or IL-6 immediately after preparation, the released cytokines that result must be synthesized by activated cells (probably monocytes), although no cause of the activation has been established (Gottschall et al. 1984). Prestorage removal of leucocytes from platelet concentrates prevents the accumulation of cytokines (Muylle and Peetermans 1994; Stack and Snyder 1994). However, the platelet has not yet been exonerated. Soluble factors associated with the platelets may play a role as well (Phipps et al. 2001).
Refrigerated storage almost certainly accounts for the lower frequency of fever observed after RBC transfusion compared with platelet transfusion. Even after refrigerated storage for 42 days, red blood cell (RBC) concentrates contain levels of IL-1 and IL-6 only slightly greater than at the time of collection, as cellular metabolism all but ceases at 4°C (Smith et al. 1993).
Effect of Rate of Infusion
It seems intuitive that when pyrogenic substances are being infused, the rate of infusion will affect the severity of the reactions. The recommendation for initiating transfusion slowly and for increasing the rate gradually to about 500 ml in 1 h in non-urgent cases rests in part on this reasoning. Clinical experience certainly supports an impression that febrile reactions are more severe when transfusions are given rapidly; however, surprisingly few data address this issue (see Grant and Reeve 1951).
Removal of Leucocytes from Red Cell and Platelet Concentrates
The majority of febrile non-haemolytic transfusion reactions can be attenuated if not prevented entirely by ensuring that red cell concentrates contain fewer than 0.5 × 109 leucocytes (Perkins et al. 1966). These levels can be achieved by centrifugation and removal of the buffy coat. Red cells that have been frozen in glycerol, thawed and washed contain only 2% of the original number of leucocytes and are unlikely to cause febrile transfusion reactions (Chaplin et al. 1956). However, highly sensitized patients may require more rigorous treatment of red cells. In a cohort of 82 thalassaemic patients receiving chronic transfusion therapy, more than one-half experienced febrile reactions following treatment with buffy coat-reduced red cells and more than one-half of all transfusions resulted in fever. Adoption of filtration methods to remove additional leucocytes (to <5 × 106 per unit) reduced both figures below 5% (Sirchia et al. 1994). Yet even leucocyte reduction to these levels does not reduce the number of febrile reactions for certain patients. In a multicentre randomized study of 531 patients with HIV infection and AIDS, 3864 red cell units were administered during 1745 transfusion episodes. Fever occurred in 16.8% of transfusions and prestorage leucocyte reduction had no effect on overall rates of elevated temperature (Lane et al. 2002). No explanation has been offered for the apparent heightened sensitivity of these patients to transfusion.
Removal of leucocytes from platelets reduces febrile reactions, but this technique is most effective when performed immediately following collection (see above). Although bedside filtration effectively removes leucocytes from platelet concentrates, controlled studies fail to demonstrate a reduction in either febrile reactions or severe reactions in the recipients (Williamson et al. 1994). Significant reductions in both febrile reactions and all categories of moderate and severe reactions have been documented when fresh platelets and platelets that are leucocyte reduced before issue are used (Enright et al. 2003).
The various filters used to remove leucocytes from red cell or platelet concentrates are discussed in Chapter 13.
In summary, for patients who have had previous transfusion reactions and in whom leucocyte-poor red cells are indicated, blood depleted of white cells by most methods will generally be effective. Commercial leucoreduction filters are most efficient in this regard, and many countries have adopted universal leucoreduction. As febrile reactions increase the patient’s oxygen requirement, complicate diagnosis and cause the patient distress, and as many patients will suffer repeated reactions (Kevy et al. 1962), the common practice of delaying the use of leucocyte-reduced components until several reactions have been experienced should be abandoned.
The Effect of Drugs in Suppressing Febrile Reactions
Antipyretics are used extensively for treatment and prophylaxis of febrile transfusion reactions. Although generations of clinicians have endorsed their use, primarily for fever related to red cell transfusion, the effectiveness of antipyretics (if any) may vary depending upon the specific blood component transfused and the mechanism of fever production. Pre-medication for platelet concentrate transfusion has not been particularly successful in preventing chills and fever, whether related to cytokine accumulation, bacterial contamination, or leucocyte antibodies (Heddle et al. 1993; Morrow et al. 1993; Wang et al. 2002; Sanders et al. 2005; Kennedy et al. 2008). However, when volunteers were given intravenous pyrogens and aspirin (1 g) was administered at the onset of shivering, symptoms were suppressed within 20 min in all but the most severe cases; moreover, there was no subsequent rise in temperature (Dare and Mogey 1954). In a single case in which a subject was given 1 g of aspirin 1 h before the injection of 10 ml of anti-D-sensitized red cells, followed by three doses of 1 g at 2-h intervals, the expected febrile reactions failed to occur (Jandl and Tomlinson 1958). For severe rigors, especially with granulocyte transfusions, intravenous meperidine 25–50 mg is remarkably effective. The mechanism of action is unknown. For less threatening reactions, paracetamol (acetaminophen) is still widely used in the treatment and prophylaxis of febrile reactions. Compared with aspirin, corticosteroids and non-steroidal anti-inflammatory agents, paracetamol does not interfere with platelet function and has a more benign adverse event profile. None of these agents appears to be particularly effective.
No compelling evidence suggests that antihistamines prevent febrile reactions, although the use is widespread. A randomized, placebo-controlled study of highly selected oncology patients could not show that oral paracetamol 650 mg and intravenous diphenhydramine 25 mg prevented any of an assortment of non-haemolytic transfusion reactions in recipients of leucoreduced single-donor platelets (Wang et al. 2002). Kennedy et al. (2008) published similar experience. In separate studies, when 5 mg of diphenhydramine hydrochloride, or 10 mg of chlorpheniramine maleate, were added to 500 ml of blood at the time of transfusion, the recipients were as likely to develop febrile reactions as those receiving untreated blood (Wilhelm et al. 1955; Hobsley 1958).
Transfusion-Related Acute Lung Injury
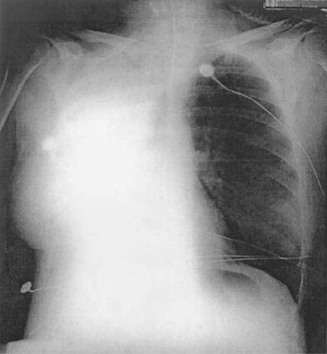
A severe transfusion reaction, originally termed non-cardiogenic pulmonary oedema, has been associated with leucocyte antibodies in donor plasma. The typical reaction is characterized by chills, fever, a non-productive cough, dyspnoea, cyanosis and hypotension or hypertension occurring within an hour or two of transfusion (Popovsky and Moore 1985). Characteristic chest radiograph findings include bilateral fluffy pulmonary infiltrates, numerous, predominantly perihilar opacities and infiltration of the lower lung fields without cardiac enlargement or engorgement of the vessels. Unlike pulmonary oedema associated with circulatory overload (TACO, Chapter 2), central venous pressure and pulmonary wedge pressure are not elevated with transfusion-related acute lung injury (TRALI). The diagnosis still rests on clinical criteria, and though several ‘consensus definitions’ have been proposed, the occurrence of respiratory distress within 6 h of initiation of transfusion, the absence of signs of circulatory overload, and radiographic evidence of new bilateral pulmonary infiltrates should be sufficient data to raise a high level of suspicion. There is no diagnostic assay. Measurement of peptides such as BNP and NT-BNP, secreted by the myocardium in response to ventricular volume and pressure distention have been proposed as a method to distinguish between TRALI and TACO, however they do not appear to be sufficiently specific, perhaps because hypoxia itself can stimulate their secretion (Li et al. 2009; Luo et al. 2006). Brittingham and Chaplin (1957) first described the syndrome and the ability to provoke it by injecting leucoagglutinins into a recipient. Fifty millilitres of blood containing leucoagglutinins with a titre of 256 were transfused to one of the two authors, who developed several of the above-mentioned signs and symptoms. Almost half a century later, an unusual opportunity to confirm the role of antibody specificity arose when a 34-year-old woman who underwent a single lung transplant developed pulmonary infiltrates in the transplanted lung following transfusion of leucocyte-reduced red cells containing anti-HLA-B44 in the plasma (Figure 15.2). The donor lung expressed the HLA-B44 antigen, but the native lung did not (Dykes et al. 2000).
Figure 15.2 Chest radiograph of a patient who received a single lung transplant and developed TRALI in the transplanted lung following red cell transfusion. The native lung is clear on the radiograph.
(Source: Dykes et al. 2000. Reproduced with permission of John Wiley & Sons Ltd.)
Several hundred cases of the classic ‘pulmonary’ reaction, now called TRALI, have been reported; the condition is clearly underdiagnosed and underreported. A review of the charts of 36 evaluable recipients of plasma from one donor whose plasma containing anti-5b (anti-HNA-3a) was implicated in a fatal pulmonary reaction, found evidence of seven mild-to-moderate and eight severe pulmonary reactions (Kopko et al. 2002; Silliman et al. 2003). Only two of the eight severe transfusion reactions had been reported to the hospital transfusion service, and only 2 out of the 15 reactions were known to the blood collector. Serious lung injury following transfusion represents only the tip of a respiratory iceberg. Many patients with unexplained pulmonary insufficiency and severe hypoxaemia, as well as others who present with few of the characteristic signs and symptoms suffer from TRALI. Some 15% of cases present with mild to moderate hypotension, typically unresponsive to fluid challenge, whereas another 15% present with hypertension (Popovsky and Haley 2000; Silliman et al. 2003). The British ‘Serious Hazards of Transfusion’ (SHOT) initiative has listed TRALI among the most common causes of transfusion-associated major morbidity and death since 1996 (Williamson et al. 1999). Mortality is estimated at 6–10% (Silliman et al. 1997; Wallis 2003).
In the US, TRALI has been estimated to occur in about 1:5000 blood component transfusions. Calculations from the British SHOT data and from the German Paul Ehrlich Institute estimate frequency at 1:65 000 FFP infusions, the highest risk component associated with TRALI. Other European haemovigilance studies report a lower incidence (Andreu et al. 2002). The true frequency of the syndrome is complicated by the combination of passive reporting and differing diagnostic criteria.
TRALI has been observed following transfusion of most plasma-containing blood components and risk appears related in part to the volume of plasma transfused. The single exception appears to be solvent detergent-treated plasma where pooling of 12 000 units during manufacture may dilute even high-titre antibody present in any single unit. As little as 2 ml of plasma seems to be sufficient to cause respiratory distress. In most cases the responsible antibodies are found in the donor. However, TRALI may also occur when the antibodies are present in the recipient and granulocytes are transfused (Gans et al. 1988; Stroncek 1996). The pulmonary infiltrates and respiratory compromise may persist for 24 to 48 h. The clinical expression and severity of disease probably owe a great deal to patient-specific factors as illustrated by a 22-year-old man with severe aplastic anaemia at NIH who experienced TRALI on two separate occasions after the inadvertent infusion of aphaeresis platelets from the same donor who was found subsequently to be alloimmunized to the human neutrophil antigen (HNA)-3a. Upon lookback, eleven other platelet components from this donor were transfused prior to that second event; two caused reactions: one chills and one TRALI, whereas nine other infusions did not (Muniz et al. 2008). In a prospective case control study, recipient risk factors identified by multivariate analysis included higher interleukin-8 levels, liver surgery, chronic alcohol abuse, shock, higher peak airway pressure while being mechanically ventilated, current smoking history, and positive fluid balance (Toy et al. 2011).
Pathogenesis of TRALI
Antibodies directed against numerous leucocyte antigens have been implicated in TRALI, including HLA antibodies (Andrews et al. 1976; Popovsky and Moore 1985; Gans et al. 1988; Eastlund et al. 1989; Kao et al. 2003), granulocyte-specific antibodies [anti-NA2 (Yomtovian et al. 1984); anti-NB2 (van Buren et al. 1990) and anti-5b (Nordhagen et al. 1986; Kopko et al. 2002)]. Dooren and co-workers (1998) reported a 31-year-old previously healthy man who suffered a severe pulmonary reaction after a 10-ml infusion of an experimental intravenous gamma globulin concentrate containing monocyte-reactive IgG antibodies. An intradonor leucocyte–antibody reaction in a patient receiving multiple platelet units has also been associated with TRALI (Virchis et al. 1997). In 20–30% of TRALI cases, no leucocyte antibody is detected. Absence of antibody may mean that the syndrome has an alternative cause (see below) or that the culprit is an antibody not detected by current methods, an important consideration as therapeutic monoclonal antibodies are used increasingly in the clinic.
The exact mechanism of TRALI, and therefore the essential characteristics of the antibodies responsible for it, remains uncertain. HLA class I, class II, and HNA antibodies have all been implicated. The latter seem to be most likely to result in severe reactions, and sensitive assays for HLA antibodies are likely to implicate many donors who pose no risk to transfusion recipients. Components with high titre HLA class II antibodies probably pose an intermediate risk to recipients with the cognate antigen. In an in vitro model in which isolated rabbit lungs and 5b antibodies were used, severe vascular leakage could be induced, but only in the presence of rabbit complement (Seeger et al. 1990). This finding implies that activation of complement is essential for inducing TRALI. Experimental models suggest that TRALI occurs when a host, with a primed immune system, is exposed to an activating agent such as a leukocyte antibody or a biologic response modifier such as lysophosphatidylcholines. Platelets may play an important role in antibody-based experimental models (Gilliss and Looney 2011). A similar mechanism in patients implicates bioactive lipids capable of priming neutrophil degranulation has been proposed (Silliman et al. 1997). In a prospective study of TRALI, Silliman and co-workers (2003) observed 90 instances of TRALI in 81 patients, a frequency of 1 in 1120 components transfused. HLA antibodies were detected in only 3.6%. Significant associations were found with increasing length of platelet storage, increased leucocyte priming activity, lipid priming activity and IL-6 concentration. In a retrospective analysis of 10 patients with TRALI, each had an antecedent ‘first event’ such as surgery, sepsis cytokine administration or massive transfusion (Silliman et al. 1997). These authors propose a ‘two-hit model’ that might explain antibody-negative cases of TRALI as well as the absence of clinical reactions in some susceptible patients who receive passively transfused antibody. However, it is clear that antibody alone can effect TRALI in an otherwise healthy subject (Dooren et al. 1998).
Prevention and Treatment of TRALI
Several strategies have been proposed to prevent TRALI. Many investigators recommend disqualification of donors associated with a case of TRALI, although most will reinstate such donors if further study detects no evidence of an antibody to a leucocyte antigen. In a randomized study, Palfi and co-workers (2001) report that plasma from multiparous women is more likely to impair pulmonary function in intensive care patients. Several countries have introduced policies that preferentially use plasma from male donors for transfusion and send plasma from female donors, and particularly those who are multiparous, for fractionation. Haemovigilance data suggest a reduction in the number of cases of TRALI following institution of such policies. More controversial are policies to screen plasma and platelet donors for evidence of alloantibodies to HLA and granulocyte-specific antigens; Screening assays have not been standardized and these methods are currently neither sensitive nor specific as regards TRALI deterrence. Washing red cell components will remove both antibody and lipid priming activity; however, such a recommendation, even for major surgical procedures, seems premature.
The treatment of TRALI requires prompt and vigorous respiratory support. Oxygenation and mechanical ventilatory assistance are often necessary (Popovsky et al. 1992). Plasmapheresis to remove the offending antibody and corticosteroids to reduce the inflammatory response have been recommended on occasion; however, little evidence supports their routine use. Patients who recover appear to suffer no residual pulmonary impairment.
Graft-Versus-Host Disease
Transfusion-associated graft-versus-host disease (TA-GvHD) occurs when immunocompetent allogeneic lymphocytes in a blood component engraft in the recipient, proliferate and mount an attack against the host tissues. The earliest reports of what was once thought to be a rare and invariably fatal disease involved children with immunodeficiency syndromes. The clinical picture evoked comparison with the runt syndrome developed by newborn mice that were challenged with adult splenocyte infusion (Hathaway et al. 1965). TA-GvH disease is probably underdiagnosed because the syndrome often affects patients who are already severely ill. The combination of symptoms may be wrongly attributed to the underlying disease, intercurrent infection or a severe reaction to a drug. Although baseline prospective studies are lacking, the frequency of TA-GvHD appears to be increasing as a result of increases in the surgical procedures, immunosuppressive therapies and transfusion strategies that predispose to allogeneic cell engraftment.
Circulating Lymphocyte Chimerism
Lymphocyte chimerism in fraternal twins, in transplant recipients and (bidirectional) in maternal–child pairs has long been recognized, but only relatively recently has the extent of chimerism following blood transfusion been appreciated (Schechter et al. 1972; Lee et al. 1999). Although the detailed fate of transfused lymphocytes is unknown, Lee and co-workers (1999) followed the kinetics of transfused lymphocytes in 10 otherwise immunocompetent trauma victims who received multiple units of red cells stored for two weeks or less. Seven of the 10 patients had evidence of multilineage (CD4, CD8, CD15, CD19) cell proliferation and circulation for up to 1.5 years; none developed GvHD.
Features of TA-GvHD
The main features of TA-GvHD involve a complex of fever and rash, nausea, vomiting and watery or bloody diarrhoea, hepatitis, lymphadenopathy and pancytopenia. Bone marrow aplasia differentiates TA-GvHD from the GvHD that follows allogeneic bone marrow transplantation. The dermatological features usually begin as a central erythematous, maculopapular eruption, which spreads to the extremities and may progress to generalized erythroderma and the formation of bullae. The histological features of most affected tissues, although not pathognomonic, are sufficiently typical, so that skin biopsy, once considered, proves an easy, sensitive and relatively benign course to early diagnosis.
TA-GvHD disease occurs between 4 and 30 days after transfusion. When the full-blown syndrome is observed and multiple organ systems are involved, mortality is high, approaching 90% (von Fliedner et al. 1983; Juji et al. 1989). However, less severe disease, especially if transient, may not be recognized, much less reported. The postoperative erythroderma syndrome reported primarily in Japan is one such example (Ito et al. 1988). TA-GvHD has been observed after transfusion of whole blood, packed red cells, platelet concentrates, granulocyte concentrates and fresh plasma, but not after fresh-frozen plasma or cryoprecipitate (Anderson and Weinstein 1990). Fresh blood may predispose to lymphocyte engraftment, although ‘freshness’ may be no more than a surrogate marker for the number of immunocompetent lymphocytes in the component.
Factors That Predispose to TA-GvHD
TA-GvH disease has been reported in six clinical circumstances:
Diagnosis and Prevention of TA-GvHD
The diagnosis of post-transfusion GvH disease can be made when the appropriate clinical picture is supported by skin biopsy; diagnosis is confirmed by detection of donor DNA. A technique based on the analysis of polymorphisms associated with variations in the length of dinucleotide or trinucleotide microsatellite repeats has been described (Wang et al. 1994).
In order to avoid the risk of GvHD from blood transfusion, the transfused components must be irradiated to inactivate donor T lymphocytes. T-cell responses become undetectable only after exposure to at least 25 Gy (Pelszynski et al. 1994). This is the dose commonly recommended in the US and in Japan; other recommendations range from 15–50 Gy. Prevention has been approached by both targeted and global irradiation strategies. Irradiation of cellular components has been advised for patients (1) with congenital immune deficiencies; (2) undergoing high-dose chemotherapy; (3) with Hodgkin disease and for (4) intrauterine transfusions; (5) transfusions for premature infants; and (6) HLA-matched platelet transfusions (Petz et al. 1993; Benson et al. 1994; Pelszynski et al. 1994) and for ‘fresh’ components (stored for 3 days or less). However, some oncology units and paediatric hospitals have elected to irradiate all blood components, and in Japan, blood component irradiation is universal. Photochemical treatment of blood components to reduce transmission of infectious agents may have the added benefit of protection against TA-GvHD (Grass et al. 1999).
Data from a mouse model suggest that as few as 107 lymphocytes per kilogram can induce the syndrome. Although methods are available to prepare blood components with such low levels of leucocyte contamination, removal of leucocytes from blood components by centrifugation or filtration has not been proven to prevent TA-GvHD. Leucocyte reduction alone is therefore not recommended as a measure to prevent TA-GvHD (see Anderson 1995).
Treatment for TA-GvHD ranges from difficult to dreadful. Survivors remain sufficiently few as to merit publication, such as a 61-year-old woman who developed TA-GvHD after treatment for chronic lymphocytic leukaemia and who then underwent intensive immunosuppression and return of autologous peripheral blood progenitor cells (Hutchinson et al. 2002). A variety of new approaches based on recent information regarding pathogenesis have been reviewed (Williamson 1998).
Reactions Due to Platelet Antibodies
Febrile Reactions
The role of platelet antibodies in causing transfusion reactions is difficult to assess, first because suspensions of platelets are always contaminated to some extent with leucocytes, and second because platelet alloantibodies are usually accompanied by leucocyte antibodies. Fever resulting from reactions to platelet-specific antibodies must be uncommon; however, Kroll and co-workers (1993) reported six cases of fever in patients with antibodies and post-transfusion purpura, who were unsuccessfully transfused with platelets. Fever, rash and thrombocytopenia were also observed in patients who were infused with plasma containing platelet-specific antibodies (Ballem et al. 1987). The observation that febrile reactions may be associated with the accumulation of cytokines in stored platelets complicates interpretation of fever in the presence of platelet antibodies. There is no doubt that the destruction of platelets by other alloantibodies can cause adverse reactions. The association was shown convincingly by Aster and Jandl (1964). HLA-A2-negative recipients were transfused with HLA-A2-positive platelets and, a day or two later, with serum containing anti-HLA-A2. In three out of the four recipients, frontal headache developed at 30 min and rigors at 45–50 min (lasting 15–30 min), followed by fever.
Post-Transfusion Purpura
In this uncommon syndrome, some patients with platelet-specific alloantibodies develop profound thrombocytopenia between 2 days and 2 weeks after a transfusion. The clinical syndrome is characterized by purpura, epistaxis, gastrointestinal bleeding and haematuria. If untreated, the course may be mild and self-limited, lasting as little as a few days or as long as several months. However, haemorrhage can be dramatic, and even when treated, mortality is reported as 5–10% (Kroll et al. 1993). It is important to distinguish this disorder from the more common syndromes of heparin-induced thrombocytopenia and thrombosis (HITT) and drug-related thrombocytopenic purpura, as the clinical course, management and outcome are quite different (Lubenow et al. 2000).
In the first recorded example of post-transfusion purpura (PTP), a platelet-specific antibody was detected and named anti-Zwa (van Loghem et al. 1959), but only later was the antibody recognized as the cause of thrombocytopenia. Shulman and co-workers (1961) described two women who developed severe generalized purpura with thrombocytopenia 6–7 days after transfusion; in both, a platelet antibody termed anti-PlA1 (later shown to be identical with anti-Zwa) was present. The investigators postulated that these two patients had been immunized to PlA1 previously during pregnancy, and that the subsequent transfusion of PlA1+ platelets had stimulated an anamnestic response. The mechanism by which the patient’s native PlA1– platelets were destroyed was (and remains) more problematic. The authors suggested that sufficient PlA1 antigen from transfused platelets circulated at least 1 week after transfusion, interacted with anti-PlA1 and somehow caused the non-specific destruction of the patient’s own PlA1-negative platelets (Shulman et al. 1961) (see below). However, the authors did not succeed in demonstrating residual PlA1 antigen in their patients. (PlA1/Zwa has now been renamed HPA-1a; see Chapter 13).
Almost all PTP patients are women; most appear to have been immunized by previous pregnancies. In a few cases, men, or women who have either never been pregnant or who had only HPA-1a-negative children, have been immunized by a previous transfusion; one case has been described in a woman without a recognized previous pregnancy or transfusion (Nicholls et al. 1970). Of the several hundred reported cases, only five were men, four of whom had been transfused many years before the transfusion implicated in PTP (Shulman 1991).
In almost all cases, the platelet-specific antibodies responsible for PTP are directed against antigens located on the GPIIb–IIIa complex. In a Joint European Study comprising 104 cases, anti-HPA-1a was found in 88, anti-HPA-1b in five, anti-HPA-3a in six and anti-HPA-3b in one case (Mueller-Eckhardt 1991). Antibodies against an antigen on GPIa–IIa (anti-HPA-5b) were found in one case (Christie et al. 1991). In another case the alloantibody anti-Naka (GPIV) was detected, but thrombocytopenia in this case may not have represented PTP (Bierling et al. 1995).
Anti-HPA-1a has usually been found in its highest titre on about the seventh day after transfusion and in many cases has disappeared completely within the following month. However, in one case the antibody persisted at 50% of its peak level after 12 months (Lau et al. 1980) and, in another, remained detectable for 18 months. The antibody was IgG alone in six out of eight cases and a combination of IgM and IgG in the remaining two. The IgG antibody was IgG1 alone in five cases and IgG1/IgG3 combined in three others. In most cases, the antibody activated complement (Pegels et al. 1981).
Pathogenesis of Post-Transfusion Purpura
The mechanism of destruction of the patient’s own platelets in PTP remains unknown. The following hypotheses have been proposed:
Immune Complex Absorption
Shulman and co-workers (1961) suggested that HPA-1a antigens present in the donor blood form immune complexes with anti-HPA-1a in the patient or that such complexes might be released after destruction of donor platelets. In either case, the complexes might adhere to the patient’s platelets, which would subsequently be sequestered. Particulate HPA-1a antigen has been detected in stored blood from HPA-1a+ donors (Shulman et al. 1961) and an increased amount of IgG has been demonstrated on the recipient’s platelets in several cases – up to 10 times the normal amount in one case on the fourth day after the onset of thrombocytopenia (Cines and Schreiber 1979). Increased antibody was demonstrable by immunofluorescence in two cases during the acute phase of the illness (Pegels et al. 1981) and, together with IgA and IgM, in five cases, in three of which anti-HPA-1a could be eluted from the patient’s HPA-1a– platelets (von dem Borne and van der Plas-van Dalen 1985). In two cases anti-HPA-1a could be eluted from the patient’s platelets even after the platelet count had returned to normal (Taaning and Skov 1991). An argument against the immune complex theory is a lack of correlation between the titre of anti-HPA-1a and the severity of thrombocytopenia (Hamblin et al. 1985; Chong et al. 1986). However, this may be due to differences in the immunochemical characteristics of the antibodies.
One possible explanation for the adherence of immune complexes to platelets in PTP involves the interaction of specific glycoproteins on the platelet membrane and on platelet microvesicles. In almost all cases of PTP, the platelet antigens involved are found either on glycoprotein GPIIb (HPA-3a, HPA-3b) or on GPIIIa (HPA-1a, HPA-1b; HPA-4a). GPIIb and GPIIIa associate readily to form the complex GPIIb–IIIa; in fact, under experimental conditions, these proteins can be kept apart only in the absence of Ca2+ (Kunicki et al. 1981; Hagen et al. 1982; Howard et al. 1982). It is possible that fragments of HPA-1a+ platelets, if they contain GPIIb–IIIa, adhere to GPIIb–IIIa on intact HPA-1a– platelets. When the platelet fragments are bound to IgG (i.e. anti-HPA-1a), the intact HPA1a– platelets with the adherent immune complexes undergo destruction by the mononuclear phagocyte system (MPS). In support of this theory is the observation that patients with Glanzmann’s disease, whose platelets lack GPIIb–IIIa, and who have antibodies against determinants on GPIIb–IIIa, have not developed PTP. In addition, two cases in which anti-HPA-5b was involved have been described; in one instance, HPA-5b was the sole antibody (Christie et al. 1991) and in the other, anti-HPA-1b was present as well (Walker et al. 1988). HPA-5b is located on GPIa, which forms a complex with GPIIa, so that the mechanism described above may be the same in these cases.
Binding of HPA-1a Antigen to Recipient’s Platelets
In vitro, HPA-1a antigen in plasma from HPA-1a+ subjects is bound by HPA-1a– platelets (Kickler et al. 1986). If plasma containing HPA-1a antigen is transfused to HPA-1a– subjects, the antigen may bind to the recipient’s platelets and thus render them susceptible to destruction by anti-HPA-1a.
Formation of Crossreacting Antibodies
It has been proposed that in the secondary immune response, in addition to anti-HPA-1a, crossreactive antibodies are formed against an epitope common to HPA-1a and -1b (Morrison and Mollison 1966). Crossreactive antibodies could not be detected, but the techniques used at that time were insensitive (see below).
Formation of Autoantibodies
Another explanation for PTP proposes the formation of autoantibodies during the secondary immune response to the alloantigen; the difference between crossreactive antibodies (see above) and autoantibodies may be more one of definition than of biology.
In animals, alloimmunization against platelets may induce the formation of autoantibodies and thrombocytopenia (Gengozian and McLaughlin 1978). In some cases, when sensitive techniques were used, serum taken during the acute phase was found to react with HPA-1a– platelets including the patient’s own platelets taken after recovery (Pegels et al. 1981; Minchinton et al. 1990). Furthermore, eluates from patients’ platelets in the acute phase reacted weakly with HPA-1a– platelets although much more strongly with HPA-1a+ platelets (von dem Borne and van der Plas-van Dalen 1985). Such reactions with HPA-1a– platelets probably result from auto-antibodies, or from crossreactive antibodies. However, in most cases acute phase serum does not react with HPA-1a– platelets and the role of autoantibodies in PTP remains unclear.
Post-Transfusion Purpura: Persisting Puzzles
One of the many perplexing features of PTP is the unpredictability of disease recurrence. No untoward episode occurred in two cases in which HPA-1a+ blood was transfused to patients who had previously suffered from PTP, even although anti-HPA-1a was still detectable in the serum. The explanation may be that some special characteristic of the antibody is required for inducing PTP. Shulman and co-workers (1961) noticed that the anti-HPA-1a antibodies in their first two patients with PTP were complement fixing in the acute phase but not in the recovery phase. Perhaps a sufficient number of immune complexes to induce PTP is released only with intravascular destruction of platelets.
PTP may recur with re-exposure years after the initial episode. An HPA-1a-negative patient who had two relatively mild episodes of thrombocytopenia separated by an interval of 3 years, both occurring a week or two after transfusion, was found to have potent anti-HPA-1a in her plasma (Soulier et al. 1979). Shulman’s original patient, who failed to develop thrombocytopenia after repeated exposure to HPA-1a+ in the months after her first episode, sustained a second, dramatic episode of thrombocytopenia following transfusion for cardiac surgery at NIH more than a decade later (NR Shulman, personal communication). Another patient had three episodes of PTP due to anti-HPA-1a (Budd et al. 1985).
A curious relationship is worth noting between PTP and neonatal alloimmune thrombocytopenia (NAIT), two different manifestations of sensitization to HPA-1a. Although 2% of the USA population lacks HPA-1a, PTP and NAIT occur less frequently than might be expected. A ‘two-hit’ mechanism has been proposed to explain this discrepancy (de Waal et al. 1986). The majority of PTP and NAIT patients express the HLA-DR3 allele, and all have the supertypic determinant DRw52. DRw52a may be the class II chain that presents the HPA-1a immunogenic peptide to antigen-specific T cells to initiate the immune response. According to this hypothesis, the majority of patients must inherit at least two specific gene loci (DRw52a and homozygous HPA-1b) to develop anti-HPA-1a and be rendered ‘at risk’ for PTP and NAIT. It is equally puzzling that among the multiparous women who have developed anti-HPA-1a and subsequent PTP, none has been reported with NAIT.
A final unexplained and as yet unconfirmed observation is the dramatic reduction in PTP that followed introduction of universal leucoreduction in the UK. Whereas leucoreduction of RBC removes most of the platelets and platelet microvesicles, reduction of platelet antigen is not a very satisfactory explanation and does not explain the decline in cases following platelet transfusion.
Management of Post-Transfusion Purpura
Many patients have been treated with corticosteroids, usually with little benefit. Administration of prednisone in doses of approximately 1–2 mg/kg per day has produced a prompt rise in platelet count in a few instances (Vaughan-Neil et al. 1975; Slichter 1982).
Exchange transfusion produced excellent responses in two of the original cases (Shulman et al. 1961; Cimo and Aster 1972) and, subsequently, many examples of rapid response to plasma exchange have been recorded. However, in at least one case the exchange of 5 litres of plasma in 3 days produced no improvement (Erichson et al. 1978).
Until recently, transfusion of HPA-1a-negative platelets to patients with established PTP due to anti-HPA-1a had been considered fruitless (Vogelsang et al. 1986; Mueller-Eckhardt and Kiefel 1988). This experience contrasts with the successful practice of transfusing antigen-negative platelets to neonates with NAIT. However, infusions of large numbers of antigen-negative platelets can effect transient increases in the patient’s platelet count and clinical haemostasis in PTP (Brecher et al. 1990; Win et al. 1995). Antigen-negative platelets, when available – usually from the patient’s relatives – can be a critical temporizing measure in emergencies. The combination of antigen-negative platelets and IVIG (see below) can reverse the course of the disease within 24 h.
Treatment with high-dose IVIG has altered the management of PTP and has become the initial treatment of choice. Following IVIG infusion, 16 out of 17 patients achieved normal platelet counts within a few days. Relapse occurred in five patients, but platelet counts returned to normal after a second dose of IVIG (Mueller-Eckhardt and Kiefel 1988). In one patient with massive bleeding, PTP was refractory to corticosteroids, IVIG and plasma exchange. An immediate and sustained rise in the platelet count followed splenectomy (Cunningham and Lind 1989). Splenectomy should be reserved for such cases.
Effect of Transfusing Platelet Alloantibodies
Experimental Studies
The effect of transfusing plasma containing platelet alloantibodies to a normal subject was described by Harrington (1954). The donor was a subject who had received many transfusions previously. Potent platelet agglutinins were found in his plasma and an injection of 10 ml into a normal subject produced profound thrombocytopenia.
Similarly, Shulman and co-workers (1961) showed that a transfusion of as little as 5–10 ml of plasma containing potent complement-fixing anti-HPA-1a would produce thrombocytopenia in normal HPA-1a+ subjects. In a subsequent review, Shulman and co-workers (1964) concluded that the concentration of alloantibody capable of producing effects in vivo was about 10 times less than the minimum concentration detectable by the most sensitive tests in vitro.
Further Studies
51Cr-labelled HLA-A2 platelets were transfused to four recipients whose own platelets lacked the antigen; 1–2 days later various amounts of serum containing anti-HLA-A2 were injected. This serum, in the presence of positive platelets, fixed complement in vitro at a dilution of 1 in 300. In the first recipient the injection of 0.25 ml of serum produced clearance of the labelled platelets with a T1/2 of 160 min and sequestration only in the spleen. In the second recipient the injection of 0.5 ml of serum produced clearance with a T1/2 of 80 min with sequestration in both liver and spleen, and the injection of 2.0 ml of serum brought about clearance with a T1/2 of 15 min and sequestration in the liver. Three of the four subjects developed febrile reactions (Aster and Jandl 1964).
Clinical Reactions
Following the transfusion of 80 ml of blood, a patient developed a severe reaction characterized by dyspnoea, rash and fever; there was marked oozing of blood and the platelet count fell from 193 × 109/l to 11 × 109/l. The donor plasma was found to contain anti-HPA-1a with a titre of 1000; the patient was HPA-1a+. Three previous recipients of blood from the same donor had developed unexplained thrombocytopenia (Scott et al. 1988). In a similar case, a patient developed severe thrombocytopenia immediately after receiving a unit of red blood cells that contained strong anti-HPA-1a (Ballem et al. 1987). Compared with six reported cases involving passive transfer of anti-HPA-1, milder thrombocytopenia has been reported following infusion of a unit of FFP that contained anti-HPA-5b; this observation may be related to the lower number of GPIa–IIa molecules on the platelet surface (2000) than with GP IIb/IIIa (60 000) (Warkentin et al. 1992).
Granulocytopenia Following Transfusion of Incompatible Platelets
In two patients with aplastic anaemia, the transfusion of HLA-incompatible platelets was followed by a prolonged decrease in the number of circulating granulocytes. At 20 h, the count was still only 30% of the pre-transfusion value and remained depressed at 48 h. No rebound granulocytosis, as noted after the transfusion of incompatible red cells, was found. HLA-matched platelets did not cause granulocytopenia (Herzig et al. 1974). The authors discussed the possibility that the transfusion of incompatible platelets may increase the risk of infection.
Reactions Due to Transfused Proteins
Immediate-Type Hypersensitivity Reactions Following Plasma Transfusion
Following the transfusion of plasma-containing blood components, the recipient may develop an anaphylactic-type reaction; the severest form (anaphylactic shock) is characterized by flushing, hypotension, substernal pain, laryngospasm and dyspnoea, and the mildest simply by urticaria (hives). Common skin hypersensitivity reactions are IgE mediated. Symptoms and signs, usually pruritus, urticaria and angioedema but including a wide range of cutaneous eruptions, have been attributed to histamine release. Severe reactions also appear to be IgE mediated, but involve a variety of chemical mediators including fragments of complement components, C3a, C4a and C5a, tryptase, cytokines, leukotrienes and platelet activating factor (Kemp and Lockey 2002).
Many cases of anaphylactic shock are attributed to an interaction between transfused IgA and class-specific anti-IgA (Chapter 14) in the recipient’s plasma, but often the cause of the reaction is unknown and the number of severe reactions attributed to IgA deficiency overall is probably exaggerated.
The frequency of severe anaphylactic reactions following transfusion is very low, one case per 20 000 transfusions in one series (Bjerrum and Jersild 1971). In contrast, urticaria during transfusion is relatively common. Kevy and co-workers (1962) report the incidence at 1.1%; in another series in which even the occasional weal was counted, the incidence was approximately 3% (Stephen et al. 1955). In a retrospective analysis of 1613 transfusion reactions during a 9-year period at a single institution, allergic reactions accounted for 17%, but of these only 7% were severe, accounting for 1.7% of all transfusion reactions. A more current retrospective analysis of 93 737 aphaeresis platelet infusions reports an overall incidence of allergic reactions of 1.72% (Savage et al. 2011). In a prospective study of platelet transfusion, 8679 transfusions in 598 patients, extensive urticaria occurred during 0.4% of transfusions and in 4.4% of patients (Enright et al. 2003). Allergic reactions occurred with a frequency of 1 in 4124 blood components and 1 in 2338 transfusion episodes (Domen and Hoeltge 2003). A retrospective study of components that were split and administered to two different recipients suggests that for aphaeresis platelets at least, recipient factors appear to be more important than component factors, and certain platelet donors have an increased likelihood of being associated with a recipient allergic reaction (Savage et al. 2011).
IgA
Reactions Due to Class-Specific Anti-IgA
Several cases have been described in which the transfusion of only a few millilitres of blood has caused such symptoms as dyspnoea, substernal pain, laryngeal oedema and circulatory collapse. These patients usually have undetectable IgA and high-titre, naturally occurring class-specific anti-IgA.
In a man who had received a transfusion of plasma some years previously, a transfusion of plasma resulted in severe symptoms, including shortness of breath and substernal pain after only 10–15 ml had been given. The symptoms subsided rapidly after the transfusion was discontinued and hydrocortisone was administered (Vyas 1970). In a similar case, intense malaise, laryngeal oedema, sweating and collapse followed the transfusion of only a few millilitres of whole blood. The patient gave a history of a similar reaction after receiving an injection of intramuscular Ig a year earlier. The very slow transfusion of washed red cells produced only slight malaise. The patient’s serum contained a potent anti-IgA (titre varying from 64 to 1000) (Ropars et al. 1973).
In another case, red cells that had been frozen and deglycerolized could be transfused without incident, provided that the rate was slow enough; rapid infusion produced a mild reaction. The transfusion of twice-washed red cells, cell-free plasma or plasma protein fraction (PPF) all produced a reaction. The patient’s own stored blood and blood from donors known to lack IgA was well tolerated (Miller et al. 1970). Subsequently, the same patient received RBC transfusions washed with 2 litres of saline without incident.
In yet another patient, twice-washed red cells gave only a mild reaction and five-times-washed red cells no reaction. In this patient, and in three others, anaphylactic reactions developed after giving a small amount of whole blood. In one case, symptoms developed within a few seconds. All four patients had anti-IgA titres in the range 500–16 000 (Leikola et al. 1973).
The frequency of IgA deficiency and of anti-IgA depends on the assay used. Rate nephelometry is a relatively insensitive assay and will not detect IgA levels of <7 mg/dl. Absent IgA by this assay requires further testing with a haemagglutination or ELISA assay that is sensitive to 0.05 mg/dl. Patients with IgA levels above this do not often have clinically important anti-IgA or anaphylaxis from transfusion.
Class-specific anti-IgA has been detected in 76.3% of 80 IgA-deficient patients with a history of anaphylactic transfusion reaction. Of these patients, 48 received 525 components drawn from IgA-deficient donors without clinical reaction (Sandler et al. 1994). However, the same study confirmed that sera of 1 in 1200 of 32 376 random blood donors tested negative for IgA and positive for class-specific anti-IgA. As the incidence of anaphylaxis is estimated at between 1 in 20 000 and 1 in 47 000 transfusions (Pineda and Taswell 1975), screening for class-specific anti-IgA by current methodology probably overestimates the risk to current transfusion recipients. True anaphylaxis is rare.
Reaction to IgA Following Injected Immunoglobulin
Although Ig preparations usually contain about 95% IgG, all of them contain some IgA. One serious reaction following the intramuscular administration of Ig was shown to be associated with the presence in the recipient’s serum of potent anti-IgA; other patients with weaker anti-IgA suffered no reactions (Vyas et al. 1968). Anaphylactic reactions due to anti-IgA occur more frequently after intravenous administration of Ig. Reactions have been shown to be particularly severe in patients with IgE anti-IgA (Burks et al. 1986; Ferreira et al. 1988). The routine screening for anti-IgA in patients to be treated with IVIG has been advocated (McCluskey and Boyd 1990). Simple methods for screening for anti-IgA have been described (Hunt and Reed 1990; Mohabir and Rees 1995).
Reactions Due to Anti-IgA of Limited Specificity
The term ‘limited specificity’ applies to antibodies against Am factors and subclass antibodies (i.e. anti-α1 and anti-α2). Only a few cases have been described in which anti-IgA of limited specificity seems to have been responsible for anaphylactic reactions.
In one case the patient, who had a severe erythematous rash, abdominal pain and difficulty in breathing following the transfusion of 100 ml of packed red cells from one donor, showed a rise in anti-IgA titre from 8 to 32. Subsequently, she was given 60 ml of plasma from the same donor, developed a severe hypersensitivity reaction and lost consciousness. Previously, she had been transfused with IgA-deficient plasma without reaction (Vyas et al. 1968).
Details of two further cases were as follows: The first patient was a woman whose second transfusion, given 2 years after the first, was followed by severe urticaria, hypotension and oliguria. A third transfusion given 2 years later produced a similar clinical picture accompanied by apprehension and respiratory distress. The patient’s serum contained anti-IgA of limited specificity with a titre of 256. In the second case a man, who may have been transfused previously, was given 2 units blood during operation and a third unit on the following day. After 25–30 ml had been transfused, he developed apprehension and restlessness and died 45 min later. The only abnormal finding in his serum was anti-IgA of limited specificity with a titre of 64 before transfusion and of 16 immediately afterwards (Pineda and Taswell 1975). In four patients of the phenotype A2m(−1), with normal levels of IgM in their serum, anaphylactoid reactions following transfusion were due to anti-A2m(1) in the recipient’s plasma and could be avoided by transfusion of A2m(−1) or IgA-deficient plasma (Vyas and Perkins 1976). In one of these patients the antibody had a titre of 256 and the patient twice had a serious anaphylactic reaction following transfusion of only small amounts of A2m(1) blood (Vyas and Fudenberg 1970).
Finally, in a patient who developed a life-threatening reaction with shortness of breath, itching, hot flushes, substernal pain, marked hypotension and cardiorespiratory arrest after the transfusion of 50 ml of pooled platelet concentrate, the serum contained an antibody reactive only with red cells coated with an A2m(1) protein from one particular subject; the patient subsequently received platelet concentrates from an A2m(1)-negative relative without incident (Strauss et al. 1983).
Prevention of Anaphylaxis in Patients Who Lack IgA
Patients who have suffered anaphylactic transfusion reactions related to plasma proteins may be transfused safely with frozen deglycerolyzed red cells or cellular components that have been washed extensively to remove contaminant plasma (Silvergleid et al. 1977; Yap et al. 1982; Sloand et al. 1990; Toth et al. 1998). Blood services have established a registry of IgA-deficient donors. Some transfusion centres now stock fresh-frozen plasma (FFP) made from such donors.
Subjects who have sustained anaphylactic reactions with transfusion and those found to have potent, class-specific anti-IgA should be given some means of emergency identification, such as a card with the information on it or a Medic Alert bracelet (Morton et al. 2002).
Other Immunoglobulins
Antibodies to Haptoglobin
Patients with haptoglobin deficiency associated with haptoglobin IgG and IgE antibodies, and who experienced severe non-haemolytic transfusion reactions have been identified in Japan (Shimada et al. 2002). The incidence of individuals homozygous for the haptoglobin deletion has been calculated at 1 in 4000 in Japanese, 1 in 1500 in Koreans and 1 in 1000 in Chinese (Koda et al. 2000). This incidence is higher than that of IgA deficiency in Japanese, so that more attention should be paid to haptoglobin deficiency and haptoglobin antibody as the cause of transfusion-related anaphylactic reactions in Asian populations.
IgG
Immediate Reactions
Injection of early preparations of Ig modified for intravenous use commonly resulted in adverse reactions, due to the presence of either substantial amounts of aggregates or prekallikrein activator in the preparations (see below). With current IVIG preparations, such reactions are rare. However, patients with hypogammaglobulinaemia, who are especially prone to anaphylactic reactions following the administration of IgG, may develop reactions with intramuscular Ig and with the 20% subcutaneous IgG preparation. This hypersensitivity may be related to the paucity of tissue-bound Ig in these patients (Barandun and Isliker 1986).
The intravenous injection of relatively small amounts of IgG, as in vials containing anti-Rh D Ig prepared by fractionation on DEAE Sephadex, seldom causes reactions; see Chapter 12. Similarly, the commercial preparation of anti-D Ig ‘Rho-GAM’, which contains 300 µg of anti-D and, presumably, about 300 mg of IgG per 2-ml vial, although intended for intramuscular use, has been given intravenously on many occasions without producing any obvious untoward effects (W Pollack, personal communication).
Reactions Associated with Repeated Intramuscular Injections of Immunoglobulin
Among 43 normal adults (the staff of a dialysis unit) receiving 5 ml of a 165-g/l solution of Ig as a source of anti-HBs every 8 weeks, four developed either chills or local oedema or generalized urticaria. Of the 43 subjects, about 80% developed IgE antibodies against other Ig classes and about 60% showed a decrease in IgE levels that lasted for several months (Ropars et al. 1979).
Possible Role of Anti-Gm
Two cases in which anti-G1m(a) appeared to be responsible for a transfusion reaction have been described (Fischer 1964; Fudenberg et al. 1964), but these cases were reported before the role of anti-IgA was appreciated, and no investigations were made to exclude anti-IgA as a possible cause. In a more recent case, in which the role of anti-IgA was excluded, following a febrile reaction the only protein antibody demonstrable in the patient’s serum was anti-Glm(z). The antibody was remarkable in that it was IgG (unlike most Gm antibodies, which are IgM) and that it had a titre of 10 000. No tests were conducted to determine the subclass of the antibody or whether it bound complement (van Loghem and de Lange 1975).
Serum sickness-like syndromes due to anti-IgG seem to be rare. One case followed an intramuscular injection of IgG (see sixth edition, p. 631).
Related posts:
 Red cell antibodies against self-antigens, bound antigens and induced antigens
Red cell antibodies against self-antigens, bound antigens and induced antigens
 Blood grouping techniques
Blood grouping techniques
 Alternatives to allogeneic transfusion
Alternatives to allogeneic transfusion
 The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
The transfusion of platelets, leucocytes, haematopoietic progenitor cells and plasma components
 The Rh blood group system (including LW and RHAG)
The Rh blood group system (including LW and RHAG)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


