At the conclusion of this chapter, the reader should be able to: • Name and compare the three complement activation pathways. • Describe the mechanisms and consequences of complement activation. • Explain the biological functions of the complement system. • Name and describe alterations in complement levels. • Briefly describe the assessment of complement levels. • Compare other types of nonspecific mediators of the immune system, including cytokines, interleukins, tumor necrosis factor, hematopoietic growth factors, and chemokines. • Discuss the clinical applications of C-reactive protein. • Compare acute-phase reactant methods. • Analyze an acute-phase protein case study. • Correctly answer case study related multiple choice questions. • Be prepared to participate in a discussion of critical thinking questions. • Describe the principle, reporting results, sources of error, limitations, and clinical applications of the C-reactive protein procedure. The complement system displays three overarching physiologic activities (Table 5-1). These are initiated in various ways through the following three pathways (Table 5-2): Table 5-1 Three Main Physiologic Activities of the Complement System Adapted from Walport MJ: Complement, N Engl J Med 344:1058–1065, 2001. Table 5-2 Initiators of Three Complement Activation Pathways Adapted from Walport MJ: Complement, N Engl J Med 344:1058–1065, 2001. The three pathways (Fig. 5-1) converge at the point of cleavage of C3 to C3b, the central event of the common final pathway, which in turn leads to the activation of the lytic complement sequence, C5 through C9, and cell destruction (Fig. 5-2). • The classic pathway is initiated by the bonding of the C1 complex, consisting of C1q, C1r, and C1s, to antibodies bound to an antigen on the surface of a bacterial cell. • The alternative pathway is initiated by contact with a foreign surface such as the polysaccharide coating of a microorganism and the covalent binding of a small amount of C3b to hydroxyl groups on cell surface carbohydrates and proteins. The pathway is activated by low-grade cleavage of C3 in plasma. • The mannose-binding lectin pathway is initiated by binding of the complex of mannose-binding lectin and associated serine proteases (MASP1 and MASP2) to arrays of mannose groups on the surface of a bacterial cell. • Cell activation, such as production of inflammatory mediators. • Cytolysis or hemolysis, if the cells are erythrocytes. The most important biologic role of complement in blood group serology is the production of cell membrane lysis of antibody-coated targets. • Opsonization, which renders cells vulnerable to phagocytosis. The classic pathway has three major stages: When fully assembled in the correct proportions, C7, C6, C5b, and C8 form the MAC (see Fig. 5-2, inset). The C5bC6 complex is hydrophilic but, with the addition of C7, it has additional detergent and phospholipid-binding properties as well. The presence of hydrophobic and hydrophilic groups within the same complex may account for its tendency to polymerize and form small protein micelles (a packet of chain molecules in parallel arrangement). It can attach to any lipid bilayer within its effective diffusion radius, which produces the phenomenon of reactive lysis on innocent so-called bystander cells. Once membrane bound, C5bC6C7 is relatively stable and can interact with C8 and C9. The biological functions of the complement system fall into the following two general categories: 1. Cell lysis by the membrane attack complex (MAC) 2. Biological effects of proteolytic fragments of complement The first category is the situation in which the MAC leads to osmotic lysis of a cell. The second category encompasses other effects of complement in immunity and inflammation that are mediated by the proteolytic fragments generated during complement activation. These fragments may remain bound to the same cell surfaces at which complement has been activated or may be released into the blood or extracellular fluid. In either situation, active fragments mediate their effects by binding to specific receptors expressed on various types of cells, including phagocytic leukocytes and the endothelium (Table 5-3). Table 5-3 Selected Complement Components and Functions Low levels of complement suggest one of the following biological effects: • Complement has been excessively activated recently. • Complement is currently being consumed. • A single complement component is absent because of a genetic defect. Specific component deficiencies are associated with a variety of disorders (Table 5-4). Deficiencies of complement account for a small percentage of primary immunodeficiencies (<2%), but depression of complement levels frequently coexists with SLE and other disorders associated with an immunopathologic process (Box 5-1). Table 5-4 Complement Deficiency in Human Beings Modified from Colten HR, Rosen FS: Complement deficiencies annual review of immunology, 10:809-834, 1992 and Nusinow SR, Zuraw BL, Curd JG: The hereditary and acquired deficiencies of complement, Med Clin North Am 69:487-504, 1985. 1. Deficiency of the opsonic activities of complement 2. Any deficiency that compromises the lytic activity of complement During immune complex reactions, certain complement proteins become physically bound to the tissue in which the immunologic reaction is occurring. These proteins can be demonstrated in tissue by appropriate immunopathologic stains. The most frequent evaluation of complement is by serum or plasma assay (Table 5-5). Complement components (e.g., C3 and C4) can be assessed by nephelometry. These assays are useful for the diagnosis and monitoring of patients. Table 5-5 Interpretation of Complement Activation by Individual Components The procedures discussed next can be used in diagnostic immunology. The factor B component is consumed by activation of the alternative complement pathway. Assessment of C3PA indicates whether a decreased level of C3 results from the classic or alternative pathways of complement activation. Decreased levels of C3 and C4 demonstrate activation of the classic pathway. Decreased levels of C3 and C3PA with a normal level of C4 indicate complement activation via the alternative pathway (Table 5-5). In SLE, both the classic and alternative pathways are activated.
Soluble Mediators of the Immune System
The Complement System
Activity
Responsible Complement Protein
Host Defense Against Infections
Opsonization
Covalently bonded fragments of C3 and C4
Chemotaxis and leukocyte activation
C5a, C3a, and C4a; anaphylatoxin leukocyte receptors
Lysis of bacterial and mammalian cells
C5-C9 membrane attack complex
Interface Between Innate and Adaptive Immunity
Augmentation of antibody
C3b and C4b bound to immune complexes and to antigen
Responses
C3 receptors on B cells and antigen-presenting cells
Enhancement of immunologic memory
C3b and C4b bound to immune complexes and to antigen; C3 receptors on follicular dendritic cells
Disposal of Waste
Clearance of immune complexes from tissues
C1q; covalently bonded fragments of C3 and C4
Clearance of apoptotic cells

Pathway
Initiators
Classic
Immune complexes
Apoptotic cells
Certain viruses and gram-negative bacteria
C-reactive protein bound to ligand
Alternate
Various bacteria, fungi, viruses, or tumor cells
Mannose-binding lectin
Microbes with terminal mannose groups
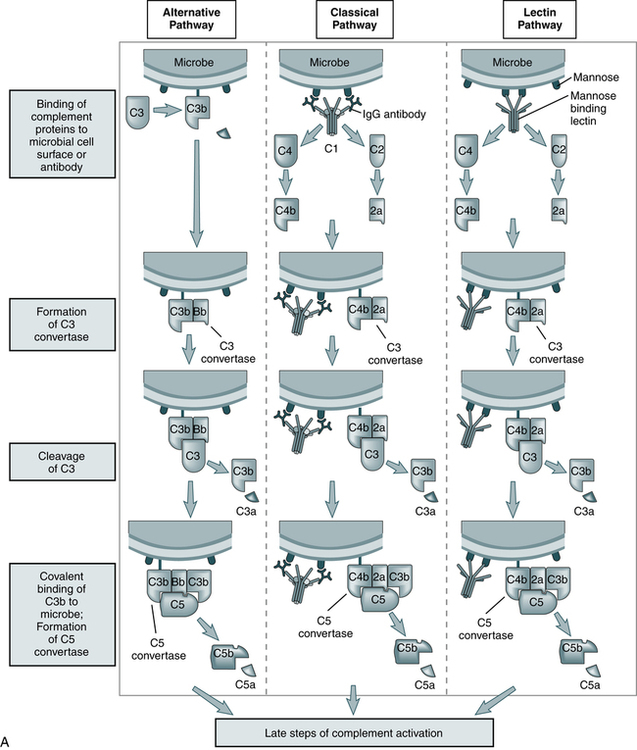
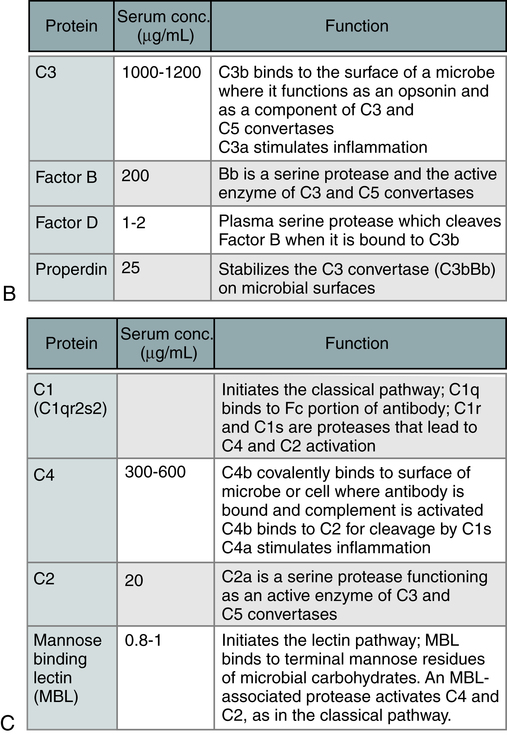
The steps in the activation of the alternative, classical, and lectin pathways are shown. Note the sequence of events is similar in all three pathways, although they differ in their requirement for antibody and in the proteins used. (From Abbas AK, Lichtman AH: Basic immunology: functions and disorders of the immune system, updated edition, ed 3, Philadelphia, 2011, Saunders.)
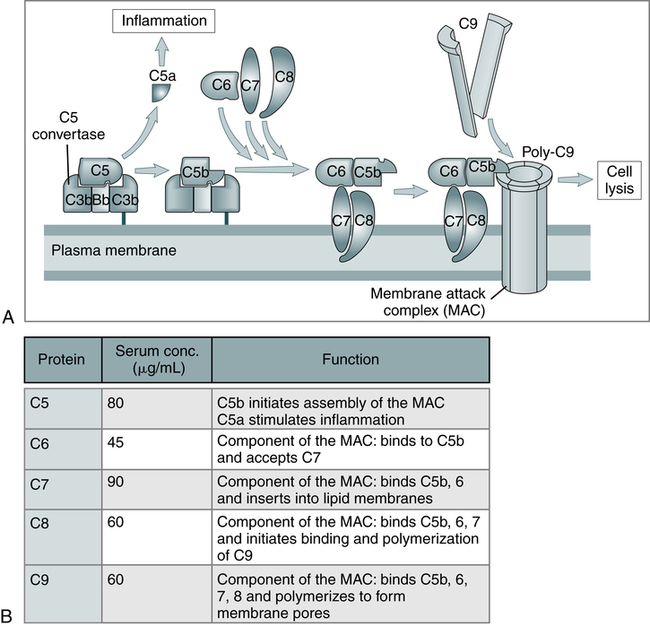
A, The late steps of complement activation start after the formation of the C5 convertase and are identical in the alternative and classical pathways. Products generated in the late steps induce inflammation (C5a) and cell lysis (the membrane attack complex [MAC]). B, The properties of the proteins of the late steps of complement activation are listed. (From Abbas AK, Lichtman AH: Basic immunology: functions and disorders of the immune system, updated edition, ed 3, Philadelphia, 2011, Saunders.)
Activation of Complement
Effects of Complement Activation
Classic Pathway
Membrane Attack Complex
BiologicAl Functions of Complement Proteins
Complement Component(s)
Function
C5-C9
Lysis of cells
C3B, IC3B
Opsonization in phagocytosis
C5A >C3A >>C4A
Anaphylatoxins/inflammation (vascular responses)
C5A
Polymorphonuclear leukocyte activation
Classic complement pathway, C3B, ?iC3b, C3dg
Immune complex removal
B-lymphocyte activation
Alterations in Complement Levels
Decreased Complement Levels
Deficiency
Associated Disease
C1q
SLE-like syndrome; decreased secondary to agammaglobulinemia
C1r
SLE-like syndrome; dermatomyositis, vasculitis, recurrent infections and chronic glomerulo-nephritis, necrotizing skin lesions, arthritis
C1s
SLE, SLE-like syndrome
C1 INH
Hereditary angioedema, lupus nephritis
C2
Recurrent pyogenic infections, SLE, SLE-like syndrome, discoid lupus, membranoproliferative glomerulonephritis, dermatomyositis, synovitis, purpura, Henoch-Schönlein purpura, hypertension, Hodgkin’s disease, chronic lymphocytic leukemia, dermatitis herpetiformis, polymyositis
C3
Recurrent pyogenic infections, SLE-like syndrome, arthralgias, skin rash
C3 inactivator
Recurrent pyogenic infections, urticaria
C4
SLE-like syndrome, SLE, dermatomyositis-like syndrome, vasculitis
C5
Neisseria infections, SLE
C5 dysfunction
Leiner’s disease, gram-negative skin and bowel infection
C6
Neisseria infections, SLE, Raynaud’s phenomenon, scleroderma-like syndrome, vasculitis
C7
Neisseria infections, SLE, Raynaud’s phenomenon, scleroderma-like syndrome, vasculitis
C8
Neisseria infections, xeroderma pigmentosa, SLE-like syndrome
Diagnostic Evaluation
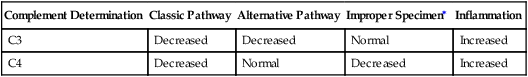
Complement Determination
Classic Pathway
Alternative Pathway
Improper Specimen∗
Inflammation
C3
Decreased
Decreased
Normal
Increased
C4
Decreased
Normal
Decreased
Increased
Assessment of Complement
C3PA (C3 Proactivator, Properdin Factor B)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Soluble Mediators of the Immune System