Fig. 13.1
Adult-type fibrosarcoma. Fibrosarcoma has evolved from among the most common diagnoses among soft tissue sarcomas to a diagnosis of rigorous exclusion. The morphology depicted, that of a “herringbone” pattern of intersecting fascicles of spindle cells with scant cytoplasm and spindled nuclei, can be demonstrated by a number of malignancies, ranging from malignant peripheral nerve sheath tumors, synovial sarcoma, cellular desmoid fibromatosis, fibrosarcomatous dermatofibrosarcoma protuberans, or low-grade fibromyxoid sarcoma, to even nonmesenchymal sarcomatoid carcinoma. The case illustrated above was a “bona fide” adult-type fibrosarcoma where synovial sarcoma, fibrosarcomatous dermatofibrosarcoma protuberans, and other entities were rigorously excluded by history, molecular assays, and immunohistochemistry
This was not always the case: fibrosarcoma was once the most frequently diagnosed soft tissue sarcoma among adults, diagnosed in over half of cases. However, due to the tripartite developments of changing classification of soft tissue lesions, emergent understanding that the histologic pattern of fibrosarcoma subtended morphologically and molecularly distinct neoplasms, and the ability to employ molecular diagnostics to recognize simulants of fibrosarcoma, this entity has become rare indeed. In fact, the author of the aforementioned review directly tested this question, recently [4]. Reviewing >150 lesions diagnosed as fibrosarcoma at the Mayo Clinic between 1960 and 2008, Bahrami et al. used contemporary nosologic criteria, immunohistochemistry, and fluorescence in situ hybridization (FISH) to reclassify each case. The diagnosis of adult-type fibrosarcoma was retained in only 16 % of cases, with the remainder reclassified as either undifferentiated pleomorphic sarcoma (20 %), specific types of fibrosarcoma (12 %), soft tissue tumors of other types presenting fibrosarcomatous histomorphologic pattern (48 %), or nonmesenchymal tumors (4 %). The most prevalent specific diagnoses, beyond the 20 % of cases of undifferentiated pleomorphic sarcoma, were synovial sarcoma (13 %), appreciated by its characteristic molecular rearrangement, solitary fibrous tumor (9 %), and malignant peripheral nerve sheath tumor (5 %). Nonmesenchymal malignancies simulating fibrosarcoma included spindle cell melanoma (3 %) and sarcomatoid carcinoma (2 %).
A Word on the Goals and Organization of This Chapter
In keeping with the spirit and theme of this textbook, focusing content to be clinically oriented, this chapter is focused on the clinical problems encountered with these lesions, presenting the types of lesions and their diagnostic settings where the tests are currently in use or likely to be implemented in the near future. Provision of an exhaustive tally of all recurrent genetic abnormalities seen in soft tissue lesions (which include both recurrent as yet not understood chromosomal translocations or regional alterations as well as some examples where specific genes have been defined) is beyond the scope of this chapter and duplicative of the very substantial efforts of the WHO Blue Book itself [1] as well as several excellent recent reviews providing a broader perspective [2, 5–9]. Instead, this chapter focuses more on clinically relevant differential diagnoses in these lesions, highlighting the surgical and molecular pathologist’s role not only in securing diagnostic confirmation but also in providing perspective on the diagnostic settings where these tests are used in a contemporary, judicial, and resource-conscious manner.
Within the framework of this clinical focus, the organization of the chapter maintains the same configuration of the current World Health Organization “Blue Book” classification of soft tissue lesions [1]. In the WHO classification, the first major branchpoint in classification of soft tissue lesions is based on their pattern of mesenchymal differentiation (e.g., adipocytic, fibroblastic/myofibroblastic, or myogenic). Then, within each differentiation pattern-based category, there are subclassifications based on risk, defining lesions as (1) benign; (2) intermediate (which includes lesions that may be either locally aggressive or even rarely metastasizing); or (3) malignant.
Indications for Molecular Testing
In an age of ever-increasing attention to value conscious practice of medicine, including in surgical pathology, it bears consideration that molecular testing of soft tissue neoplasms is not an end in and of itself: use of molecular testing must add some value to the management of an individual patient’s soft tissue lesion [10]. The broad question of indications for molecular testing has been reviewed in detail recently [2]. The most common indication for molecular testing, anticipated by the case study of fibrosarcoma, above, is for confirmation of the diagnosis when multiple entities may present similar morphologies. Let there be no misunderstanding in this regard; lesions showing morphology within the morphologic spectrum of “spindle cell sarcoma,” especially on small biopsies, may vary from benign reactive processes (e.g., the wide variety of pseudosarcomatous myofibroblastic proliferations) to benign neoplasms (e.g., nodular fasciitis), to intermediate, locally aggressive but not metastasizing, processes (e.g., desmoid-type fibromatosis), to intermediate, rarely metastatic processes (e.g., dermatofibrosarcoma protuberans), to fully malignant tumors of varying grades (e.g., low-grade fibromyxoid sarcoma or adult fibrosarcoma). The prototypical example in surgical pathology where multiple entities present similar morphology is that of tumors with “small round blue cell” morphology, where lesions varying from high-grade neuroendocrine (small cell) carcinoma, high-grade hematopoietic malignancies such as lymphoma or leukemia, and high-grade round cell sarcomas present an essentially identical histologic appearance. Each of these varied entities requires use of specific and differing multimodal therapeutic interventions.
The importance of securing appropriate classification of soft tissue neoplasms, and by its extension, the utility of molecular diagnostics for confirmation, is underscored by the grading systems used for soft tissue sarcomas, which include the (US) National Cancer Institute (NCI) System and the French Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), reviewed recently [11–13]. Both of these systems lean heavily on assessment of the histologic type of tumor, whether principally or indirectly. The NCI system is directly based on the tumor histology, modifiable somewhat based on other factors [14], whereas the FNCLCC system assesses a composite score based on tumor differentiation, mitotic count, and tumor necrosis that translates into a three tiered system based on a sum of the scores for a tumor evaluated across these parameters [15]. However, the histological type of the tumor enters strongly into the calculus, as the histologic type or variant determines the differentiation score that goes into the total, which ranges from a score of 1 for lesions such as well-differentiated liposarcoma to a score of 2 for less differentiated lesion such as myxofibrosarcoma or leiomyosarcoma, to a score of 3 for most pleomorphic sarcomas. As noted by the recently revised WHO classification of soft tissue tumors [1], the most widely used grading system for soft tissue sarcomas is the FNCLCC system, which seems to have some advantages among adult soft tissue sarcomas [16]; the use of this system going forward underscores the importance of assessment proper histologic classification, and by extension, establishes molecular diagnostics as a key player in prognostication.
Moreover, soft tissue pathology is one area where differential diagnoses for a given morphology—fibrosarcomatous morphology for instance—are widely different depending on the clinical scenario, including the patient sex, age, physical exam findings, and imaging characteristics. Knowledge of age- and site-specific differential diagnoses allow the practicing surgical pathologist to know that a lesion with fibrosarcomatous morphology arising in the deep soft tissues of an extremity of an infant is likely to represent infantile fibrosarcoma, a lesion with characteristic t(12;15), NTRK3-ETV6 gene fusion, which carries a favorable prognosis if completely excised [17]. Based on this clinical scenario, the pathologist knows that diagnostic workup must exclude embryonal rhabdomyosarcoma, which can arise in the same age group and location, but instead requires surgery with multimodal adjuvant chemoradiotherapy [18]. Similarly, a lesion with fibrosarcomatous morphology arising in retroperitoneum of an adult requires exclusion of its most likely explanation, dedifferentiated liposarcoma, while a lesion based in the subcutaneous tissue of the trunk of a young adult requires consideration of fibrosarcomatous transformation of dermatofibrosarcoma protuberans. In contrast, all bets are off when one of these lesions arises in an uncharacteristic anatomic location or demographic group—a scenario presenting a second general indication for use of molecular testing.
Another key indication for molecular testing in soft tissue lesions concerns the phenomenon of dedifferentiation, where tumors may progress to lose their characteristic pattern of dedifferentiation. The archetypical example of this process is that of the well differentiated liposarcoma/atypical lipomatous tumor to dedifferentiated liposarcoma sequence, where a recognizable lesion of adipocytes progresses in a time-dependent manner to a high-grade sarcoma demonstrating any one of a number of patterns of differentiation [19] and where molecular testing for the characteristic amplifications of chromosome 12q13–15 locus of MDM2 can be very helpful in establishing diagnosis. Another example includes the recently described phenomenon of dedifferentiation in solitary fibrous tumors [20, 21]. In both cases, an abrupt transition is observed between the recognizable well differentiated and dedifferentiated components such that a high-grade, dedifferentiated sarcomatous overgrowth is seen juxtaposed to and arising from the well differentiated component.
Lastly, direction of therapeutic choice remains one of the most compelling indications for molecular testing in the area of soft tissue tumors—where not only does the molecular test result drive therapy by establishing the diagnosis but where the molecular test is predictive of response to the therapy. The quintessential case where this is true is that of gastrointestinal stromal tumor (GIST), where individual mutation types of the receptor KIT or PDGFRA define populations responsive to various receptor tyrosine kinase inhibitors [22]. While the examples of such direct, predictive, molecular tests remain limited in current clinical practice, it is greatly hoped that additional opportunities to use molecular insights to personalize therapy by directly targeting oncogenic signaling pathways will come online in the near future [23, 24].
Adipocytic Tumors
Adipocytic tumors represent a class of lesions showing a dizzyingly variable morphology but are defined by “lipomatous” or adipocytic differentiation—differentiation toward that of mature fat cells. This group of tumors represents the “bread and butter” of soft tissue pathology, ranging from the most banal of benign tumors, like lipomas, the most common mesenchymal neoplasm of adults, to emerging and controversial entities, like spindle cell liposarcoma [25–27], which has not yet been well-characterized and recognized as an entity by the WHO. More importantly, the diagnostic feature that defines these lesions, observation of areas of lipomatous differentiation, may be only focally apparent or apparent to an extent such as may be present (as a metaplastic phenomenon) in other non adipocytic mesenchymal tumors. For a complete review of the spectrum of adipocytic tumors and their clinicopathologic features, we recommend the WHO Blue Book Classification [1], or any of several excellent texts [28–30].
Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma and Dedifferentiated Liposarcoma
The first clinical scenario where molecular testing may be indicated in lipomatous neoplasms concerns lesions showing morphology in the atypical lipomatous tumor/well-differentiated liposarcoma–dedifferentiated liposarcoma spectrum of tumors. Atypical lipomatous tumor is an intermediate risk lesion characterized histologically by morphologically mature adipose tissue with varying degrees of atypia. Clinically these tumors show a propensity for recurrence if incompletely resected and show an anatomic site and time dependent propensity for “dedifferentiation,” where transformation into a nonlipogenic sarcoma of varying morphology and grade is observed histologically, with concomitant acquisition of metastatic potential.
The surgical pathologist’s approach to these tumors is complicated by variable terminology applied to lesions, with different designations are given to lesions with identical morphology and molecular pathogenesis based on their anatomic location. Conventionally, when these tumors arise in peripheral sites on the trunk or on the extremities, use of the term atypical lipomatous tumor (ALT) was recommended, while for tumors arising in deep soft tissues of the thorax and abdomen, the term well-differentiated liposarcoma (WDL) was instead recommended. To emphasize, no molecular, histologic, or immunohistochemical feature distinguishes these two—they are identical lesions labeled differently depending on where they arise anatomically. The differing designations reflect the fact that tumors arising in surgically resectable somatic soft tissue sites have a risk of dedifferentiation less than 5 % and negligible overall mortality rate, while retroperitoneal tumors have a risk of dedifferentiation >20 %, with mortality as much as 80 % [31–33]. In the most recent 2013 WHO, the section heading lists only ALT; however, the authors support retention WDL as an appropriate term for tumors in locations for which margin-negative resection is nearly impossible to achieve.
The diagnostic difficulty that arises with ALT/WDL pertains to the difficulties that may arise in histologic review of lipomatous neoplasms. The histologic hallmark of ALT/WDL is observation of stromal cells with markedly enlarged and hyperchromatic nuclei seen in the setting of atypical fat (often variably sized vacuoles interspersed with unusually shaped septae and vessels) (Fig. 13.2). Lipoblasts may be observed, though they are neither highly sensitive nor specific for ALT/WDL. Unfortunately, these diagnostic cells are, often only identified widely scattered in a background of relatively non-atypical, lipoma-like neoplastic fat, which may show varying features insufficiently distinctive from normal or non-adipose neoplasms.
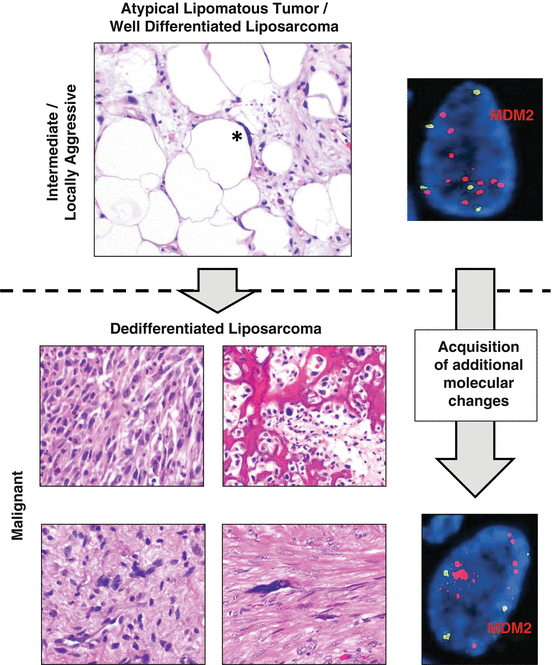
Fig. 13.2
The spectrum from atypical lipomatous tumor to dedifferentiated liposarcoma. Atypical lipomatous tumor (ALT), a locally aggressive soft tissue neoplasm, is the most common type of liposarcoma. The alternate designation of “well-differentiated liposarcoma” may be used for these tumors arising in locations where clear margins are unlikely to be secured surgically, with ALT preferred in more superficial sites where complete resection is possible. The histologic hallmark of ALT is markedly enlarged, atypical hyperchromatic stromal cells (asterisked, upper panel) seen in a background of better differentiated adipose tissue. As a time-dependent phenomenon, these tumors may undergo dedifferentiation into a nonlipogenic malignant sarcoma of varying grades and morphology, designated dedifferentiated liposarcoma (DL). DL may show a wide variety of appearances, including undifferentiated pleomorphic (upper left), spindle cell (bottom left), heterologous osteosarcomatous (upper right), or leiomyosarcomatous (lower right). Whether ALT or DL, the molecular hallmark of this sequence of tumors is amplification of the MDM2 locus at 12q14–15, which is generally present as supernumerary ring chromosomes. These may be readily identified in archival sections by interphase fluorescence in situ hybridization (FISH) using fluorescent probes flanking the MDM2 locus (red), with comparison to green centromeric probes used as a surrogate for ploidy (green). FISH micrographs courtesy of Kevin Baden and Dr. Jean Lopategui
Moreover, histologic variants observed among ALT/WDL include the inflammatory variant, where the ALT/WDL recruits a marked, mixed inflammatory, and edematous background obscuring the nature of the lesion and simulating abscess, infection, or even pseudosarcomatous myofibroblastic lesions. Another prevalent histologic variant is the sclerosing variant, which shows scattered, often markedly atypical stromal cells in a densely collagenized sclerotic background, often with some chronic inflammation, simulating cicatrix, idiopathic (or IgG4-related) sclerosing disease, or even fibromatosis [34]. The classic scenario concerns the “lipoma-like” variant, where diagnostic atypical stromal cells might only be observed focally in one of many sections of otherwise very mature-appearing fat. Lastly, as appreciated particularly recently [35], ALT/WDL may show prominent myxoid change, which may simulate benign myxoid neoplasms ranging from lipoma and myxoma to myxoid liposarcoma (see below).
Even the clinical settings where ALT/WDL is considered provide challenges. The classic scenario concerns an abdominal or retroperitoneal mass sampled by core biopsy, showing the appearance of only mature adipose tissue, raising concern for lipoma-like WDL versus a sample of adjacent adipose tissue. For that matter, sclerosing WDL may simulate retroperitoneal fibrosis [34]. Even in more accessible sites, evaluation of large lipomas or recurrent lipomas is often rendered difficult by incomplete sampling or artifacts such as fat necrosis. In fact, four scenarios in particular are highlighted in the recently published new edition of Enzinger and Weiss’s Soft Tissue Pathology as indications justifying the use of adjunctive molecular testing in these tumors, including (1) “recurrent” lipoma; (2) lipoma greater in size than 15 cm; (3) lipomatous neoplasms with atypia that is equivocal in degree; and (4) retroperitoneal and intrabdominal lipomatous lesions, regardless of the atypia [29].
Fortunately, ALT/WDL demonstrate highly prevalent though not entirely specific molecular features, which include the classic finding of supernumerary and giant marker chromosomes containing amplified sequences from the 12q14–15 locus of MDM2 [36, 37]. MDM2, which functions as an inhibitor of p53 activity, is consistently overexpressed in this setting, as are several genes flanking this locus, most importantly, from a diagnostic standpoint, CDK4. Due to this amplification and resultant overexpression, these proteins may be detected immunohistochemically [38–40], by array-based comparative genome hybridization (aCGH) [41], or, perhaps best studied and validated, by fluorescence in situ hybridization [34, 42, 43]. These techniques represent one of the most clinically useful and important applications of molecular diagnostics in soft tissue pathology, given the prevalence of these tumors and diagnostic conundra.
Dedifferentiated Liposarcoma
Dedifferentiated liposarcoma (DL), as alluded above, represents the morphologic transformation of an ALT/WDL into a nonlipogenic sarcoma of varying grade [1, 36] (Fig. 13.2). Because the dedifferentiated component of DL may show an extraordinarily variable morphology that otherwise might be diagnostic of a wide spectrum of mesenchymal lesions, its recognition is accomplished by sampling and observing a transition between an ALT/WDL into DL. Dedifferentiation is thought to be a time-dependent phenomenon, based on the fact that it is generally observed with higher prevalence among ALT/WDL of deep sites, particularly the abdomen and retroperitoneum in patients middle age and older, with equivalent prevalence in males and females. Dedifferentiation is observed most frequently at the time of diagnosis, though it may also occur in the site of local recurrence of an ALT/WDL (including ALT/WDL incorrectly diagnosed as “lipoma” or other benign entity). As with ALT/WDL, the prognosis of DL is driven by anatomic site of occurrence, which in turn modifies the lesion’s resectablility. Overall, however, the prognosis is substantially worse, such that local recurrence is observed in ~40 % of cases, metastasis in ~25 % of cases, and disease-specific mortality in ~30 % cases [33, 44, 45].
Much of the difficulty of diagnosis of DL derives from the morphologic variability of this tumor and its frequent occurrence in anatomically “deep” sites limiting diagnostic sampling. Without the classic gross or imaging finding of a tumor with admixed fatty and solid components, the dedifferentiated component may simulate with remarkable fidelity the morphology of sarcomas of other lineages, including leiomyosarcoma, rhabdomyosarcoma, osteosarcoma, and undifferentiated pleomorphic sarcoma, including its morphologic variants such as myxofibrosarcoma and inflammatory undifferentiated pleomorphic sarcoma [19]. Most interesting is the more recently appreciated phenomenon where dedifferentiated liposarcoma may show morphology of “homologous dedifferentiation,” where the DL shows myxoid morphology simulating myxoid liposarcoma [35] or even enlarged markedly atypical lipoblasts of pleomorphic liposarcoma [46].
Similar to ALT/WDL, DL demonstrates amplification of the chromosome 12q locus of MDM2 with resultant overexpression of this gene and flanking sequences, including CDK4 [36]. Similar to ALT/WDL, overexpression of MDM2 and CDK4 may be detected immunohistochemically [38–40], by array-based comparative genome hybridization (aCGH) [41], or interphase FISH performed on archival sections [34, 42, 43]. Quite interesting and controversial, in terms of application of this technique, has been the issue of peripheral undifferentiated pleomorphic sarcomas harboring MDM2 amplification. Such cases, to present understanding, would be morphologically and molecularly indistinguishable from dedifferentiated liposarcoma arising “de novo” without a detectable well-differentiated component. Emerging data, including one well-designed clinicopathologic and molecular study, make a convincing argument that they show, similar to DL, superior prognosis to other undifferentiated pleomorphic sarcomas [47]. Whether such a potential prognostic application of MDM2 FISH is of any role in undifferentiated pleomorphic sarcoma is controversial, but may be contemplated given increased efforts to attempt to subclassify such tumors, going forward [48].
Myxoid Liposarcoma (Myxoid/Round Cell Liposarcoma)
Myxoid liposarcoma is a myxoid malignancy characterized by a proliferation of primitive, round to spindle cell mesenchyme admixed with a myxoid stroma containing a variable number of lipoblasts (Fig. 13.3). The lipoblasts most characteristically show a “signet right cell”-type morphology with univacuolation; however, variable amounts of the tumor may show greater differentiation with lipomatous areas which may render the differential with lipoblastoma or ALT difficult. Most distinctive, and diagnostically useful, is the characteristic vasculature of this sarcoma; these tumors demonstrate a vasculature of fine arborizing capillaries with a “chicken-wire” appearance. The stroma shows most characteristically a prominent myxoid appearance, often with areas of pooling of mucins, imparting sometimes cystic or microcystic appearance; the quality of myxoid pools of these areas has been described as “pulmonary edema-like”. In some areas, the adipocytic component may show microvacuolation, imparting the appearance of brown fat or hibernomatous differentiation [49, 50]. Historically, high-grade myxoid liposarcomas were designated “round cell liposarcoma”; however, the most recent WHO has removed this term “round cell” as the hypercellular and high-grade areas in a myxoid liposarcoma are not exclusively of round cell morphology and often show spindle cell features [1]. Definitionally, high-grade myxoid liposarcoma shows at least 5 % of a more cellular component composed of nearly solid proliferation of the round-to-spindled primitive mesenchymal cells with a sufficient degree of cellularity that a subset of nuclei appear to touch. For high-grade tumors the risk for metastatic outcome is increased; overall approximately 30–40 % of cases demonstrate metastasis, which is further correlated with large size (>10 cm) or coagulative necrosis [51, 52]. Metastatic sites may include unusual locations, including to soft tissue or bone. Myxoid liposarcoma remains the most common liposarcoma of the very young (first and second decades) but is overall a tumor of young adults, arising most characteristically in the deep soft tissues of the arms and legs [36, 50, 52].
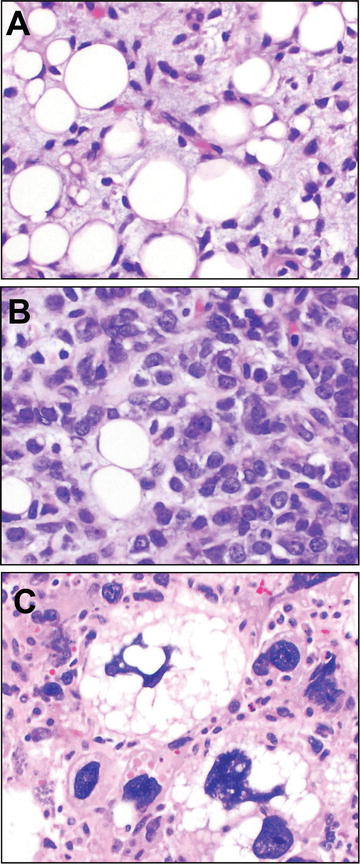
Fig. 13.3
Myxoid liposarcoma and pleomorphic liposarcoma. Myxoid liposarcoma is a malignant lipogenic sarcoma demonstrating round to ovoid primitive-appearing spindle cells admixed with univacolar signed ring lipoblasts in a myxoid stroma. The vascular pattern is described as arborizing “chicken wire” capillaries that are helpful in its recognition (a). More challenging are high-grade cases, defined as >5 % of the tumor area with dense cellularity such that the nuclei appear to touch or overlap (b). Especially in the metastatic setting these lesions can engender the classic small round cell differential, for which it is quite helpful that myxoid liposarcoma exhibits characteristic fusion of the genes FUS and DDIT3. In contrast, pleomorphic liposarcoma is defined by histomorphologic observation of atypical lipoblasts, which are markedly enlarged lipoblasts showing a hyperchromatic, pleomorphic nucleus scalloped by cytoplasmic vacuoles (c). These are often seen scattered in a background of cells with high-grade undifferentiated pleomorphic or myxofibrosarcomatous pattern. Pleomorphic liposarcoma demonstrates variable, complex structural chromosomal changes currently not tested in diagnostic practice, but does not harbor the MDM2 amplification of the atypical lipomatous tumor-dedifferentiated liposarcoma spectrum of tumors or the FUS–DDIT3 fusions of myxoid liposarcoma
The morphologic differential diagnosis of myxoid liposarcoma is broad and includes a number of benign and malignant entities. First, especially in small biopsy myxoid liposarcoma may simulate benign lesions, such as lipoblastoma, myxoid dermatofibrosarcoma protuberans, intramuscular myxoma, or low-grade fibromyxoid sarcoma, especially among younger patients. In older patients, ALT or even DL with myxoid change enters the differential, while in older patients myxofibrosarcoma quickly enters the differential. Lastly, metastatic high-grade myxoid liposarcoma with predominant or exclusive round cell morphology may raise the entire classic carcinoma versus hematopoietic versus round cell sarcoma differential.
For these reasons, it is very helpful that myxoid liposarcoma shows a prevalent (>90 %) and characteristic genetic feature, translocation of chromosomes 12 and 16 resulting in t(12;16) and fusing the genes FUS and DDIT3 [53, 54]. Less frequently, EWSR1, the Ewing Sarcoma locus, another highly promiscuous fusion partner in sarcomas, may be involved, producing a t(12;22) EWSR1-DDIT3 fusion gene [55]. This translocation has been tested most frequently through use of FISH; break apart at the DDIT3 locus has been used, as has FISH for FUS or even EWSR1 [56]. While DDIT3 rearrangement using break apart FISH probe is more specific to myxoid liposarcoma, as compared to other lesions, in appropriate differential diagnostic considerations FUS or EWSR1 break apart FISH probe may be helpful but present in other lesions in the differential. Certainly, RT-PCR-based strategies designed to detect the fusion gene product show attractive sensitivity with a high degree of specificity to the lesion [57].
One final emerging consideration in lipomatous lesions concerns the diagnosis of the neoplasm, spindle cell/pleomorphic lipoma. These tumors are among the most frequent diagnostic challenges in soft tissue pathology and are frequently seen in consultation [58]. These are neoplasms of older individuals, arising on the trunk or extremities, characterized by a variable admixture of non-atypical spindle cells and mature adipocytes arrayed in a myxoid stroma with “ropey collagen” and mast cells [59]. The pleomorphic lipoma version of this tumor includes, admixed with the adipocytes and bland spindle cells, a proliferation of atypical-appearing multinucleate tumor giant cells with peripherally oriented nuclei, so-called floret cells [60]. Thus, it is not surprising that the differential diagnostic considerations depend on whether a lesion shows predominant spindle cell lipoma or pleomorphic lipoma morphology. For the spindle cell variant, considerations include myxoid liposarcoma, myxoid dermatofibrosarcoma protuberans, superficial acral fibromyxoma, low-grade fibromyxoid sarcoma, peripheral nerve sheath tumors, and myxoid solitary fibrous tumor, which may show lipomatous differentiation [61]. For lesions with predominantly pleomorphic lipoma morphology, ALT, particularly with sclerosing morphology, enters the differential. From the standpoint of molecular testing, these lesions probably bear most consideration from the exclusion of lesions in the differential; for instance, FISH or RT-PCR-based detection of characteristic genetic lesions of myxoid liposarcoma, ALT, dermatofibrosarcoma protuberans (see below), or solitary fibrous tumor (see below). One emerging area concerns the recent detection of prevalent, recurrent chromosomal loses at 13q and 16q [62]. Most usefully, the locus at 13q includes the locus of the cell cycle regulatory and tumors suppressor gene, RB, which may be detected as loss of immunohistochemical expression of RB protein [63, 64]. This has proven quite useful, including recent findings suggestion that these tumors have a very close molecular relationship with mammary-type myofibroblastoma and cellular angiofibroma (a fibroblastic/myofibroblastic tumor) [62, 64, 65].
One emerging caveat includes a controversial, still poorly defined class of lesions called spindle cell liposarcoma, which show many similar features to spindle cell lipoma but are distinctive for nuclear atypia in the spindle cell component that is beyond allowable for spindle cell lipoma. At least preliminarily, these tumors also show loss of the RB gene by FISH, which would suggest that immunohistochemistry as a surrogate for molecular features would not exclude this lesion from spindle cell lipoma [27]. In contrast, another emerging entity, “fibrosarcoma-like lipomatous neoplasm,” with histologic features in a similar spectrum has been recently described, though this lesion seems to lack loss of genetic material at the 13q locus of spindle cell lipoma, as well as the characteristic MDM2 amplifications of ALT and translocations of myxoid liposarcoma [25]. One can envisage that clarification of the nosologic and prognostic relationships between these lesions will be forthcoming.
Fibroblastic/Myofibroblastic Tumors
Fibroblastic and myofibroblastic tumors include a broad range of tumors of varying biologic potential. This area is one of particular challenge, as the degree of simulation between neoplasms in this category is remarkable, as well as is the degree of simulation between lesions arising in a reactive setting and bona fide neoplasms. For that matter, emerging data are identifying recurrent genetic abnormalities even in lesions that have been previously considered to represent reactive changes, resulting in changing their classification. In this class of lesions, the clinical setting where molecular testing is indicated most frequently is for classification of a spindle cell lesion showing a challenging morphology overlapping between entities or for a suboptimal biopsy or excision where diagnostic features are not present. Because these lesions all are fibroblastic to myofibroblastic spindle cell lesions in a similar spindle cell differential, each entity will be covered sequentially.
Nodular Fasciitis
Nodular fasciitis is a perennial problem in surgical pathology because it shows clinical and morphologic features that often appear quite worrisome for sarcoma [66]. Arising characteristically among young adults reporting rapid growth of a subcutaneous soft tissue mass, these lesions, composed of stellate spindled myofibroblasts (SMA positive in tramtrack pattern) in a loose, arcuate, or feathered “tissue culture” distribution, with, depending on the duration of the lesion, variable cellularity, myxoid change/myxoid cystic areas, or densely sclerotic collagenized areas (Fig. 13.4). In particular, the “burnt out” sclerotic appearance (with keloidal-type collagen fibers) is more prevalent in lesions excised latter in the disease course. Most pertinent to indications for molecular testing, these tumors may show frankly alarming mitotic rates and cellularity, though atypical mitotic figures are not seen. The observation of extravasated stromal red blood cells is very prevalent (but nonspecific), while osteoclast-type giant cells and chronic inflammation may be identified. From a prognostic standpoint, these lesions are benign and require diagnostic separation from intermediate (inflammatory myofibroblastic tumor, fibromatosis) or malignant (leiomyosarcoma, myofibroblastic sarcoma, low-grade fibromyxoid sarcoma, or myxofibrosarcoma) processes [67].
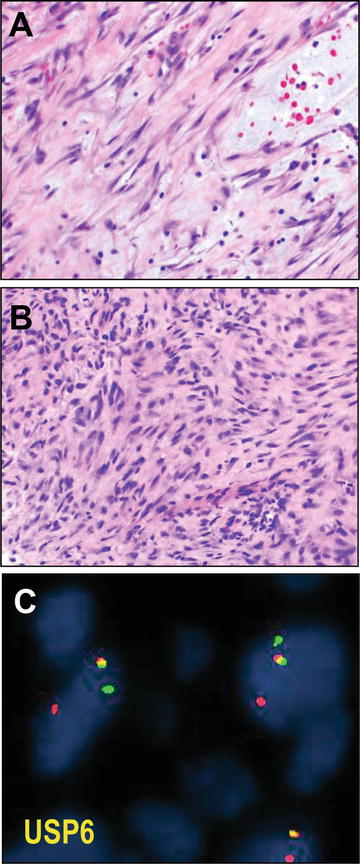
Fig. 13.4
Nodular fasciitis. Nodular fasciitis is a neoplasm of myofibroblasts demonstrating a variably cellular (a versus b) proliferation of spindle cells arranged in curved, intersecting fascicles imparting a so-called “tissue culture” appearance. Often areas appear torn with stromal mucin pools; extravasated blood is a helpful diagnostic feature (a, upper right). Confirmatory of the neoplastic nature of these self-limited tumors, and helpful diagnostically for cellular lesions which may appear worrisome (such as b), these tumors harbor rearrangement at the USP6 locus (fusing it with MYH9), which may be tested by red and green fluorescence in situ hybridization (FISH) probes flanking the locus that if intact appear to overlap (yellow) and if rearranged are split (c). USP6 FISH micrographs courtesy of Dr. André Oliveira
Fortunately, nodular fasciitis has been recently confirmed as a clonal neoplasm, harboring a reproducibly identified fusion of the genes, MYH9 and USP6. This fusion results in induction of the full length USP6 deubiquitinating protease transcription product under the MYH9 promoter [68]. Most remarkable, then, is that, in comparison to other cases of neoplasia related to gene fusions, nodular fasciitis is a benign and self-limited neoplasm nearly always without recurrence even after incomplete excision. In any case, USP6 interphase break apart FISH strategies have been devised and are now in clinical use, at least by reference labs, for diagnosis of nodular fasciitis (see Fig. 13.4). One suspects that use of this assay will even become even more widespread, given the recent observation of similar USP6 fusion genes in primary (but not secondary) aneurysmal bone cysts [69].
Fibromatosis
Fibromatosis, writ large, includes several lesions. These include the benign but clinically troublesome proliferations of palmar fibromatosis (Duputren disease) and plantar fibromatosis (Ledderhose disease), both classified as superficial fibromatoses, and the inflammatory/sclerosing lesions of penile fibromatosis (Peyronie disease) [1]. In contrast are the clearly neoplastic, locally aggressive myofibroblastic lesion of desmoid-type fibromatosis. Desmoid fibromatoses are tumors that may occur over the full lifespan, showing predominance in females during young adulthood, with predilection for the abdominal wall. Among children and older adults, equally in both sexes, desmoids show predilection for the limb girdles, head and neck, and chest wall [70]. The abdominal examples arise classically from the abdominal wall, especially the rectus muscles, or deeper sites such as the mesentery. Similar to the superficial fibromatoses, the treatment for desmoid fibromatoses, whether abdominal or extraabdominal, is surgical resection, but with wide margins in the case of desmoid tumors. These tumors show expression of muscle-specific actin and smooth muscle actin, the latter in a myofibroblastic “tramtrack” pattern, while being negative for S100 or desmin, as would have been expected for smooth muscle or neural neoplasms that they may occasionally simulate.
Most difficult in the diagnosis for these tumors is the differential between them and either reactive lesions (pseudosarcomatous myofibroblastic proliferations, including inflammatory myofibroblastic tumors), or well-differentiated leiomyosarcomas, low-grade fibromyxoid sarcomas (Evans tumors), or sclerosing lesions (including IgG4-related lesions such as sclerosing mesenteritis). For that matter, distinguishing between desmoid fibromatoses and scar, especially in instances such as margins of resections presents a significant challenge. Fortunately, bona fide desmoid fibromatoses, in the sporadic setting, demonstrate highly prevalent mutations of the gene β-catenin (CTNNB1) [71]. In fact, in a survey of well over 250 desmoid fibromatoses and nearly 200 cases of spindle cell lesions in the differential, no mutation of CTNNB1 was detected in tumors that may simulate desmoid fibromatosis, while nearly 90 % of desmoid tumors were positive for the mutation by contemporary methods [72]. The authors noted that sequencing was possible using archival paraffin tissues, including relatively scant biopsy material, with less than 5 % of cases not able to be analyzed. In widespread use, as a surrogate marker for either CTNNB1 or APC mutation (the latter in desmoid tumors arising in the setting of FAP), is immunohistochemistry for β-catenin, which shows nuclear positivity in ~80 % of these tumors [73, 74]. Though this stain can be challenging to optimize, perform, and interpret, in selected cases it may be very helpful to support the diagnosis, or, assist in parsing perilesional reactive and scar tissue versus involvement by fibromatosis, particularly at margins (Fig. 13.5). However, it has recently been shown that recurrence potential is not related to binary, positive/negative margin status but rather to positive margins or close (<1 mm) margins [75].
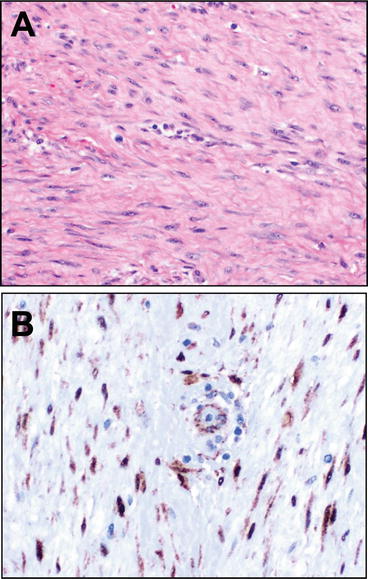
Fig. 13.5
Desmoid. Desmoid-type fibromatosis is an infiltrative neoplasm characterized by long sweeping fascicles of bland, well-differentiated spindle cell myofibroblasts with coursing through a variably, usually densely collagenized matrix (a). The nuclei lack hyperchromasia but may show mitotic activity. Helpfully, given the occasionally challenging differential with cicatrix and other reactive and neoplastic lesions, sporadic tumors harbor a mutation of the gene, CTNNB1, β-catenin, which results in nuclear accumulation of the protein (tumors associated with FAP have APC mutations, which also leads to Beta-catenin nuclear overexpression). Though mutational testing is commercially available for clinical use, these tumors are often tested via immunohistochemistry for (b), where upon careful inspection at high power the nuclear accumulation can be seen in the lesional cells but sparing admixed vascular smooth muscle or endothelium
Dermatofibrosarcoma Protuberans
Dermatofibrosarcoma protuberans (DFSP) is a locally aggressive neoplasm thought to arise from dermal fibroblasts of cutaneous sites. It is generally a neoplasm of the young, with slight male predominance, characteristically arising in the dermis of the torso, proximal limbs, and head and neck. These are cutaneous masses, often plaque-like to nodular/multinodular, notable for infiltration of the dermis and subcutaneous fat by a generally cellular, storiform proliferation of spindle cells, frequently involving subcutaneous adipose tissue with a broad invasive front and entrapping adipocytes, imparting a honeycomb pattern (Fig. 13.6). While myxoid, pigmented (Bednar tumor), and myoid variants are recognized, the storiform growth of these tumors is telltale, as is the prevalent expression of CD34. Infrequently, these tumors may show morphologic transformation to overtly sarcomatous morphology, that of “fibrosarcoma,” with concomitant acquisition, generally, of increased cellularity, atypia, mitoses, and classic “herringbone” fibrosarcomatous architecture. These tumors present significant clinical difficulty due to their infiltrative growth and resultant propensity to recur [76] if margins are not widely free [77].
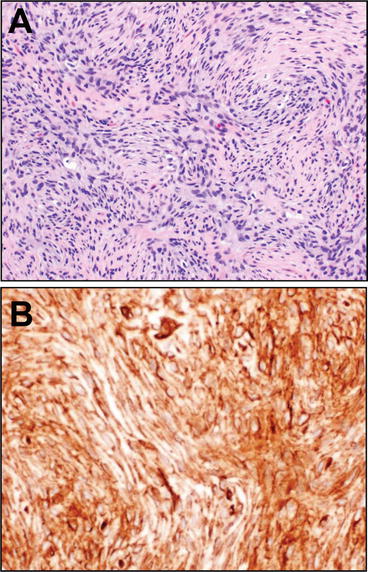
Fig. 13.6
Dermatofibrosarcoma protuberans. Dermatofibrosarcoma protuberans is characteristically a dermal- and subcutaneous-based lesion composed of bland, monomorphic spindle cells with distinctive, infiltrative, storiform growth (a). Useful, from a diagnostic standpoint, in most cases CD34 immunostain is diffusely, strongly positive (b). Thus, molecular testing for the characteristic COL1A1-PDGFB rearrangement that is characteristic of this tumor is only infrequently required from a diagnostic standpoint though may be useful in cases with fibrosarcomatous transformation
These lesions may prove diagnostically challenging for several reasons. The differential diagnosis is often with cellular benign fibrous histiocytoma, a benign so-called fibrohistiocytic lesion that also arises in the dermis, as well, occasionally, with cellular benign and malignant nerve sheath tumors (neurofibroma and malignant peripheral nerve sheath tumors) or even spindle cell lipomas, given the frequent adipose involvement. From the molecular standpoint, DFSP has long been known to harbor supernumerary ring chromosomes containing sequences of chromosomes 17 and 22 [78, 79]. More prevalent among pediatric cases are unbalanced translocations, t(17;22), while among the closely related lesion, giant cell fibroblastoma, balanced t(17;22) are mostly seen [80]. These genomic rearrangements result in fusion of the gene, α1 type I collagen preprotein, COL1AI at 17q21 to the platelet derived growth factor β-chain gene (PDGFB) at 22q13. This results in a fusion gene product composed of both exons of COL1A1 and all of PDGFB, save for the first exon [81]. Subsequent cellular processing of the fusion gene product results in elaboration of the active PDGFB growth factor ligand, which stimulates the PDGFRB receptor in DFSP cells in an autocrine manner to proliferate, resulting in induction of lesion observed clinically.
This molecular lesion may be assayed by RT-PCR [82]- and FISH-based [83] strategies currently in clinical use for diagnosis of DFSP, including its transformed fibrosarcomatous variant [84]. Not only can such assays be helpful for delineating the molecular pathogenesis of a “fibrosarcoma” where a conventional DFSP precursor lesion is not identified histologically, but these molecular assays provide an exemplar for the paradigm of molecular lesion directed therapy in soft tissue tumors: the PDGFB to PDGFBR signaling can be targeted by the small molecule kinase inhibitor active on the PDGFBR receptor, imatinib, which has been found efficacious in DFSP [85].
Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs) are a classic fibroblastic neoplasm characterized by spindle cells with small fusiform to ovoid nuclei with limited indistinct cytoplasm, coursing in a “patternless pattern” through a collagenized stroma. Classically, these lesions exhibit a so-called “hemangiopericytoma-like” vascular pattern of ramifying “staghorn” angulated vessels, a feature that was first appreciated among what is now regarded as the “cellular” variant of solitary fibrous tumors, the hemangiopericytoma described in 1942 by Stout and Murray [86]. In contrast, the fibrous, less cellular variant was described in the pleura somewhat earlier by Klemperer and Rabin in 1931 [87]. In the intervening years it became apparent that a number of non-SFT lesions were included in Stout’s original report of hemangiopericytomas, and with increasing study and tighter criteria [88], as well as exclusion of non-SFT lesions that may simulate the same morphology, these lesions became understood to represent a spectrum of the same entity [89], just as additional entities, including giant cell angiofibroma [90] and orbital fibrous histocytoma [91] have come to be included in this entity. Rendering this evolving diagnostic spectrum even more challenging is the array of morphologic variation that may be demonstrated by SFTs, which further includes myxoid, lipomatous, and cystic morphologies (Fig. 13.7).
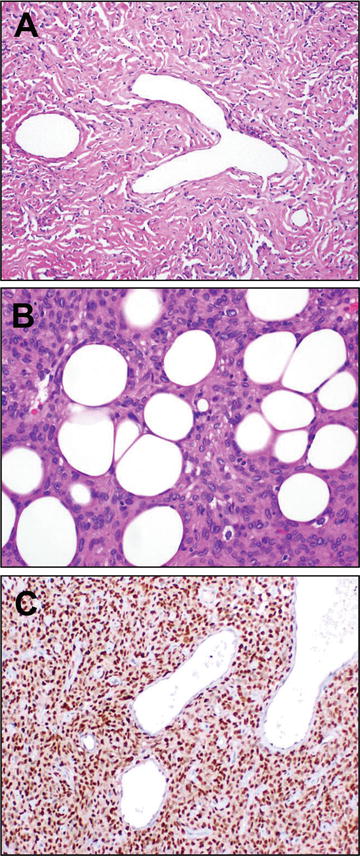
Fig. 13.7
SFT. Solitary fibrous tumor is characterized histologically by proliferation of variably cellular, bland spindle to ovoid shaped cells arranged loosely in a “patternless pattern,” intimately admixed with a collagenous stromal background. Staghorn, so-called “hemangiopericytoma”-like vessels are characteristic (a). Other cases, which may have been designated as “hemangiopericytoma” previously, show a more cellular morphology; lipomatous differentiation (b) may be seen and raise consideration of a primary lipomatous neoplasm. Identification of an intrachromosomal inversion resulting in fusion of the genes NAB2 and STAT6 has provided a unifying molecular explanation for the genesis of these tumors. Immunohistochemistry for STAT6 may be used diagnostically for specific identification of tumors in this class, where diffuse nuclear positive staining is strongly supportive in an appropriate immunomorphologic context
These are tumors generally of adults occurring over a wide age range, equally among genders, and at a wide array of anatomic sites. SFTs occur in the pleura of the lungs, where they may grow large, but also can occur in other visceral sites, the head and neck, including orbit and CNS sites, and deep soft tissue; these tumors only rarely grow at cutaneous sites [92]. SFTs usually present clinically as well circumscribed, growing masses, but also may exceptionally trigger hypoglycemia due to paraneoplastic elaboration of insulin-like growth factor. Most cases are benign, though a subset, usually with mitoses, necrosis, large size [92] or dedifferentiation [20], can be malignant and require more aggressive therapy. The classic adjunctive diagnostic feature, first adopted for use to separate pleural SFTs from malignant mesothelioma [93], is expression of CD34.
Not surprisingly, for a lesion that may show a wide array of morphologies and occur at essentially any site or age, SFTs represent a frequent diagnostic challenge. More cellular cases can simulate monophasic synovial sarcoma, dermatofibrosarcoma protuberans, true myopericytic lesions such as myopericytoma and glomangiopericytoma, and cellular benign fibrous histiocytoma. Myxoid and lipomatous lesions may simulate myxoid liposarcoma or spindle cell lipoma. Hypocellular lesions can simulate a range of tumors from calcifying fibrous tumor, to sclerosing atypical lipomatous tumor, or even fibromatosis.
For this reason, the observation via Next-Generation Sequencing strategies by two groups in 2013 of a recurrent gene fusion in solitary fibrous tumors [94, 95], of an intrachromosomal inversion fusing exons of both the genes NAB2 (transcriptional repressor) and STAT6 (transcriptional activator) provides a welcome pathognomonic molecular feature. The NAB2–STAT6 fusion, though variable in its structure, deletes part of the repressor domain of NAB2 and fuses it to the transcriptional activating domain of STAT6, thereby converting a feedback inhibitor of proliferation genes (wild-type NAB2) into a feed forward activator of proliferative genes (including the aforementioned insulin-like growth factor) that drive the neoplasm. Because this molecular lesion is a highly prevalent, seemingly universal feature of these tumors, including, as recently observed, cases arising in the central nervous system [96], a number of strategies have been used to test for it [94–96]. Interestingly, in contrast to many of the genomic rearrangements and fusion genes noted previously, FISH poses significant difficulties, because the chromosomal inversion that results in NAB2-STAT6 is relatively small and not detectable by conventional cytogenetic methods, while the final fusion oncogene is variable in its exon structure such that a sensitive RT-PCR-based assay requires a highly multiplex reaction [97]. In contrast, STAT6 immunohistochemistry has proven to be a sensitive and specific test for solitary fibrous tumor [96, 98], which is now detectable using a new monoclonal antibody [99]. Going forward, it is anticipated that this antibody will provide an invaluable addition to the growing immunohistochemical diagnostic armamentarium for soft tissue lesions.
Inflammatory Myofibroblastic Tumor
Inflammatory myofibroblastic tumor is a myofibroblastic lesion, classically of the young, of intermediate biologic potential such that recurrence is frequent but metastasis is rare [100, 101]. These tumors show a morphology of spindle to stellate smooth muscle actin positive myofibroblastic cells growing in a fascicular to storiform pattern, and admixed with a chronic inflammatory infiltrate, frequently including plasma cells. The background stroma may be variably myxoid to sclerotic and densely collagenized. They may arise across a wide variety of anatomic sites, initially characterized in the lung, causing pain and shortness of breath, and variously termed “inflammatory pseudotumor” and “plasma cell granuloma” [102]. Extrapulmonary sites are well characterized, including abdominal sites in the mesentery and omentum, as well as the gastrointestinal tract, causing obstructive symptoms [100, 101]. Examples in the urinary bladder, where they have been termed “pseudosarcomatous myofibroblastic proliferations,” “postoperative spindle cell nodule” (in the post-instrumentation setting), and pseudosarcomatous fibromyxoid tumor [103, 104], are well characterized, as well.
Interestingly, across sites, and especially among young patients, approximately half of these tumors have rearrangements at the anaplastic lymphoma kinase, ALK locus. In contrast to the gene fusions generally characterizing anaplastic lymphoma (nucleophosmin, or NPM1 fused to ALK), these fuse a number of other genes that contain oligomerization domains, including TPM3 and TPM4, as well as CLTC, CARS, and RANBP2, to a truncated kinase domain of ALK, resulting hypothesized oligomerization and aberrant, oncogenic kinase activation [105]. From a diagnostic standpoint, this feature has the potential to be quite useful. Not only is the resultant ALK expression detectable immunohistochemically, detectable in as many as 60 % of these lesions [106] but, given the widening availability of FISH for the ALK locus in light of a diagnostic and predictive role in EML4-ALK rearranged lung adenocarcinoma [107, 108], FISH has become increasingly available for these tumors (Fig. 13.8). Perhaps most importantly as a role for ALK testing is the recent observation of efficacy of the ALK inhibitor, crizotinib in IMTs [109], which could secure for recurrent or refractory cases a role for this testing in a fashion analogous to that of lung adenocarcinoma [108]. Last, it bears mention that very recent scholarship identifies multiple, potentially actionable non-ALK fusions among the remaining half of ALK rearrangement negative IMTs. Lovly et al. recently reported a Next-Generation Sequencing-based approach to study of ALK negative IMTs, finding additional prevalent kinase fusions, including ROS1 and PDGFRB [110].
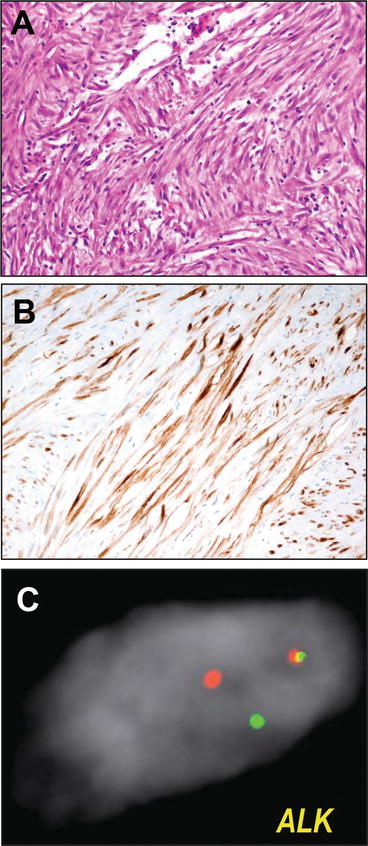
Fig. 13.8
Inflammatory myofibroblastic tumor. Inflammatory myofibroblastic tumors are characterized histologically by a proliferation of spindle cell myofibroblasts coursing through a myxoid to myxocollagenous stroma with scattered inflammatory infiltrate including plasma cells (a). Because primary myofibroblastic and smooth muscle lesions are often in the histologic differential, immunohistochemistry for ALK (b) is useful diagnostically. However, given its increasing availability and use for ALK-rearranged lung adenocarcinomas, ALK fluorescence in situ hybridization assays, using red and green fluorescent probes flanking the ALK locus to assay for rearrangements (split signals) (c) have become more widely available for diagnostic use. ALK FISH micrographs courtesy of Dr. Bryan Betz
Low-Grade Fibromyxoid Sarcoma (Evans Tumor)
Low-grade fibromyxoid sarcoma (LGFMS) is a distinctive low-grade fibroblastic sarcoma composed of bland neoplastic fibroblasts with only low-grade atypia, coursing through a variably cellular myxocollagenous stroma, often with a very zonal appearance between more collagenous and more myxoid areas [111]. A number of these cases show prominent nodules of collagen that earned a morphologic variant of this tumor the term “hyalinizing spindle cell tumor with giant rosettes” [112]. This tumor arises generally in deep soft tissues of the proximal extremities, though cases are reported at a wide variety of sites, including viscera; males appear more frequently affected than females. Despite the low-grade appearance, the behavior of these lesions can be aggressive; with long term follow-up, recurrence is seen in as much as the majority of cases, while metastasis is seen in nearly one half [113].
LGFMS presents a particularly difficult differential diagnosis, and one where this true malignancy is in a differential with lesions that are benign or relatively indolent (myxomas, myxoid neurofibroma or soft tissue perineurioma, fat-poor spindle cell lipomas [58], solitary fibrous tumors), locally aggressive (extrabdominal desmoid fibromatosis) or even malignant (low-grade myxofibrosarcoma, malignant peripheral nerve sheath tumors). For this reason, the observation of recurrent genetic lesions in these tumors provides a useful opportunity as a molecular diagnostic. These tumors show prevalent balanced translocations involving the FUS locus (or rarely EWSR1 [114]), with the genomic locus of one of two different related proteins, CREB3L2 or CREB3L1, resulting in a t(7;16) translocation fusing FUS–CREB3L2 or t(11;16) translocation fusing FUS-CREB3L1 [115–118]. For this reason, in cases that pose a diagnostic challenge, FUS rearrangement using break apart FISH probe can be used as a diagnostic adjunct. Most recently, however, gene expression profiling studies of these sarcomas and profiling expression of genes induced by transformation of cells with the fusion transgene [117] identified the protein, MUC4, as a characteristic and prevalent feature of LGFMS [119] (Fig. 13.8). This particular strategy highlights the use of immunohistochemistry for genes with expression patterns tightly correlated to the molecular status as surrogates for the molecular test.
Sclerosing Epithelioid Fibrosarcoma
Sclerosing epithelioid fibrosarcoma (SEF) is a rare sarcoma, described by Meis-Kindblom et al. in 1995 as a tumor showing malignant epithelioid cells in clusters to most classically cords coursing through a densely fibrotic stroma (Fig. 13.9) that usually imparts a zonal appearance with hypercellular and hypocellular areas [120]. These rare tumors, distinguished from carcinoma with cordlike growth and sclerosing morphology by lack of expression of epithelial markers, tend to arise among young patients, with a female predominance, in the deep soft tissues of the trunk or proximal extremities. These tumors tend to be aggressive, with upwards of 50–80 % showing recurrence, metastasis, and demise from disease [121]. Recent observations suggest that the aforementioned low-grade fibromyxoid sarcoma (above) [113], and its morphologic variant, hyalinzing spindle cell tumor with giant rosettes [112] may have morphologic, immunophenotypic, and even molecular overlap with a subset of SEF [114, 119, 122, 123]. Indeed, cases of LGFMS with SEF-like areas (which have been termed “hybrid LGFMS-SEF”) have been described and have shown prevalent expression of the LGFMS and FUS–CREB3L2/L1-fusion associated marker MUC4 [7].
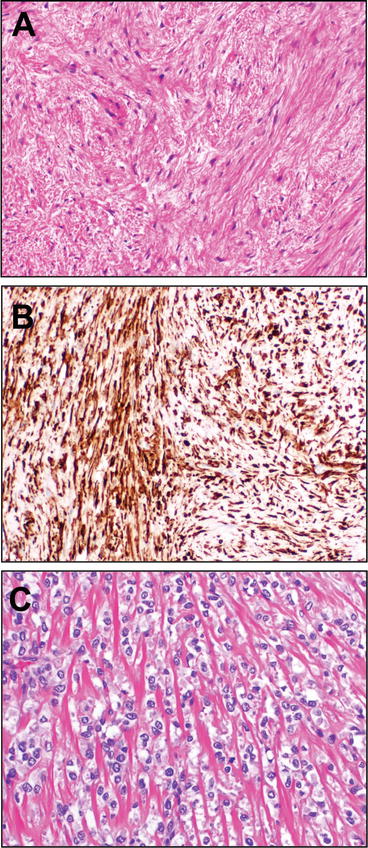
Fig. 13.9
Low-grade fibromyxoid sarcoma (Evans tumor) and sclerosing epithelioid fibrosarcoma. Low-grade fibromyxoid sarcoma is a malignant sarcoma (LGFMS), generally of the soft tissues of the extremities of young adults, showing a distinctively variable pattern of cellularity, with alternating areas of increased cellularity and areas of more myxocollagenous stroma. The spindle cells of this tumor are bland, without the telltale atypia of other myxoid sarcomas (a). Quite useful from a diagnostic standpoint was the discovery, from gene expression profiling studies of this tumor, that the FUS-CREB3L2/L1 gene fusion characteristic of this sarcoma induces expression of MUC4, which may be detected by facile immunohistochemistry (b). Very recent data have confirmed the long-postulated relationship between LGFMS and sclerosing epithelioid fibrosarcoma (SEF), which shows distinctive cords of epithelioid cells coursing through a dense fibrous stroma (c). A subset of SEFs are now known to harbor rearrangements with CREB3L2/L1, but, in contrast to LGFMS, EWSR1 is substituted for the FUS gene
Interestingly, very recent data propose the missing molecular link between these tumors, a link that is quite tractable for contemporary molecular testing. First, multiple groups have reported that EWSR1 rearrangements, substituting EWSR1 for FUS rearranged and fused with the CREB3L3/L1 loci (similar to rare cases seen in LGFMS [114]) occur in SEF [124] with high prevalence [122, 125]. Interestingly, the vast majority of pure SEF cases showed EWSR1 rearrangements, with either CREB3L1 or, less frequently, CREB3L2, while SEFs with hybrid LGFMS-SEF features showed FUS–CREB3L2 fusion [125]. These results suggest, though more study is needed, the notion that each specific type of genomic rearrangement may even be correlated to morphology. Either way, contemporary EWSR1 and FUS interphase break apart FISH methodologies are directly adoptable for diagnosis of this entity.
Skeletal Muscle Tumors
The WHO recognizes a total of five entities among skeletal-muscle tumors, including benign rhabdomyoma, embryonal rhabdomyosarcoma, alveolar rhabdomyosarcoma, pleomorphic rhabdomyosarcoma, and spindle cell/sclerosing rhabdomyosarcoma [1]. The relevance of molecular testing to this group of tumors does not apply to the first entity, which is either a histologically distinctive benign soft-tissue neoplasm showing mature skeletal muscle differentiation (adult rhabdomyoma) or a benign rhabdomyoblastic tumor showing myotube-like differentiation (fetal rhabdomyoma). Both arise most frequently in the head and neck and are associated with Gorlin Syndrome (nevoid basal cell carcinoma syndrome) with its known mutations of PTCH. In contrast, rhabdomyosarcoma represents a clinically and morphologically heterogeneous group of entities with clear molecular associations (Fig. 13.10). Discrimination between these latter malignancies is exceptionally important, given the substantially better prognosis of embryonal rhabdomyosarcoma and spindle cell/sclerosing rhabdomyosarcoma than alveolar rhabodomyosarcoma. Given the frequent occurrence of embryonal and alveolar rhabdomyosarcoma in children (while both sarcomas may also occur in adults) and the preponderance of protocol-based therapy in pediatric sarcomas, the use of molecular testing for subclassification of pediatric rhabdomyosarcomas has become de rigeur, with protocols requiring molecular confirmation even in cases where the diagnosis has been secured on morphology and immunophenotype.
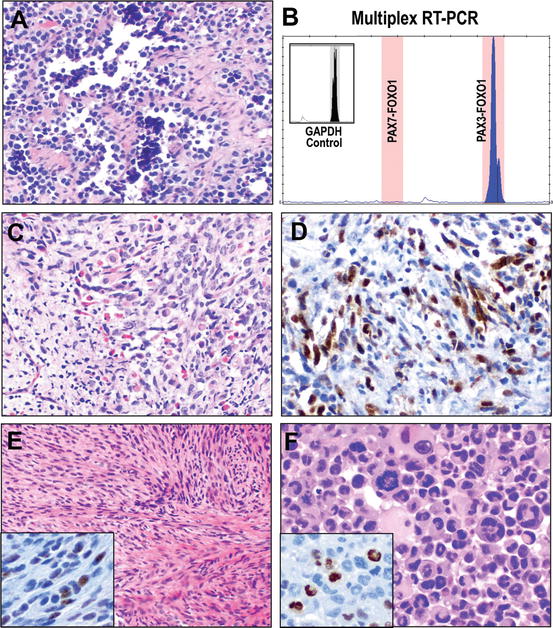
Fig. 13.10
Classification of rhabdomyosarcoma. The current WHO Classification recognizes four variants of rhabdomyosarcoma. Alveolar rhabdomyosarcoma is a poorly differentiated round cell sarcoma composed of cells with hyperchromatic nuclear atypia and scant cytoplasm arrayed classically in packeted/septate architecture imparting a pseudoalveolar appearance due to the central dyscohesion of lobules of cells (a). While this appearance raises the classic differential of “small round blue cell tumors,” the characteristic gene fusion of PAX3 or PAX7 with FOXO1 of alveolar rhabdomyosarcoma may be detected by a number of assays, including RT-PCR, as illustrated by this capillary electropherogram (x-axis, size of amplicon (bp), y-axis intensity, inset GAPDH positive control for assay and RNA quality) showing detection of expression of the PAX3-FOXO1 fusion transcript (b). In contrast, embryonal rhabdomyosarcoma demonstrates a morphology of proliferating primitive spindle cells in loose fascicles and sheets with varying proportions of rhabdomyoblasts at different stages of skeletal muscle differentiation (c). While pathognomonic gene fusions have not been detected in embryonal rhabdomyosarcomas, mutation of the gene encoding the myoid differentiation transcription factor gene, MYOD1 has been reported in approximately 10 %; whether or not it is mutated, nuclear MYOD1 expression by immunohistochemistry remains an important marker of myogenic differentiation (d). Spindle cell/sclerosing rhabdomyosarcoma has recently been recognized as a separate entity with distinct clinicopathologic features. Among adults spindle cell rhabdomyosarcoma is an aggressive malignancy, though pediatric cases, which recent data suggest may harbor NCOA2 gene rearrangements, are less aggressive. Either way, these show a cellular spindle cell proliferation in fascicles (e), though scattered foci of rhabdomyoablastic differentiation are seen and may be highlighted by immunohistochemistry for nuclear myogenin (MYF4, inset). Pleomorphic rhabdomyosarcoma shows a morphology reminiscent of undifferentiated pleomorphic sarcoma, though striking eosinophilic cytoplasm and clusters of multinucleate atypical rhabdomyoblasts are identified (f), confirmed as rhabdomyoblastic differentiation by myogenin (inset). RT-PCR electropherogram courtesy of Dr. Bryan Betz
Alveolar Rhabdomyosarcoma
Alveolar rhabdomyosarcoma (ARMS) is a high-grade sarcoma composed of malignant small round cell morphology thought to represent a very primitive population of cells with arrested myogenic differentiation. While these tumors may occur at all ages, ARMS tends to occur most frequently in a slightly older patient population than embryonal rhabdomyosarcoma, and is less prevalent [126], occurring most commonly in the extremities, genital region, paraspinal sites, or paranasal sinuses [1, 127]. The morphology of these tumors is highly cellular, composed of dyscohesive sheets of primitive round cells with a monomorphous appearance. The “alveolar” designation reflects the variable propensity for these expansile tumors to elaborate fibrous septations resulting in a nested morphology where individual sarcoma cells remain attached to the periphery of the “alveolus” and detached from the central cluster of cells (Fig. 13.10a). However, this classic appearance may be only focally present or even absent in some cases of the solid variant of ARMS [128], highlighting the difficulty of separating this tumor from cases of other small round cell sarcomas. For that matter, less frequent cases may show composite morphology with areas more similar to embryonal rhabdomyosarcoma [129].
For this reason, the identification, through classic cytogenetic means, of recurrent translocations of t(2;13) and, less frequently, t(1;13) in these tumors provides a helpful molecular correlate. The former translocation results in fusion of the gene PAX3 and FOXO1, while the latter results in fusion of the related gene, PAX7 and FOXO1 (reviewed recently [127]). Both function as transcription factors driving oncogenesis in these sarcomas [130]. Interestingly, and relevant to molecular diagnostics for these tumors, the high level of expression of PAX3-FOXO1 fusion is thought to be mostly driven by transcriptional overexpression, while PAX7-FOXO1 is upregulated by amplification [131]. Either way, these fusions may be tested by RT-PCR (Fig. 13.10b) or FISH methodologies that can either demonstrate break apart FISH at the FOXO1 locus or dual or multicolor fusion strategies demonstrating fusion of FOXO1 to either PAX3 or PAX7.
Embryonal Rhabdomyosarcoma
Embryonal rhabdomyosarcoma (ERMS) is the most frequent soft tissue sarcoma of children and adolescents, showing predilection for males, though these tumors also may occur in adults. These sarcomas show disparities in incidence by race, including a preponderance of cases arising among whites as compared to individuals of African, Latino, Native American, or Asian descent [132]. These tumors arise only infrequently in skeletal muscle; instead the head and neck and genitourinary sites are most prevalent, including the bladder, prostate, and paratestis. These tumors show a spindle cell morphology often showing variably cellular fascicles to sheets of primitive mesenchymal spindle cells with an admixture of rhabdomyoblasts showing varying degrees of differentiation (Fig. 13.10c, d). Atypical or anaplastic cells are often apparent focally. The characteristic “botryoid variant” shows subepithelial condensation of these spindle cells at mucosal sites, often imparting a polypoid lesional architecture. It is important to note, however, that dense cellular examples may show features difficult to distinguish from alveolar rhabdomyosarcoma [133], the predominant differential in this case after exclusion of other mesenchymal lesions (e.g., malignant peripheral nerve sheath tumors, especially malignant Triton tumors).
From a molecular standpoint, these tumors often show complex karyotypes with gain or loss of multiple whole chromosomes (most frequently chromosome 8) or segments. Individual genes or chromosomal loci may also show copy number gains [18]. Thus, use of FISH or RT-PCR-based strategies to exclude the characteristic translocations/gene fusions of ARMS presents the most important consideration for diagnostic testing, and which is often required by contemporary treatment protocols. However, very recently new data have been reported lending some clarity to the molecular pathogenesis of ERMS. A whole exome sequencing-based study, reported by Kohsaka et al. recently characterized a recurrent mutation of the skeletal muscle development gene, MYOD1 occurring in approximately 10 % of ERMS. The specific mutation, Leu122Arg, is hypothesized to mediate a switch between differentiation (the wild-type gene’s function) and proliferation, and be associated with a subset of cases showing lesions in the PI3K/AKT pathway [134]. Given that a number of targeted inhibitors of this pathway are in varying stages of pharmacologic development, it is anticipated that prospective molecular identification of ERMS with this aggressive molecular subtype may be of use in the very near future.
Pleomorphic Rhabdomyosarcoma and Spindle Cell/Sclerosing Rhabdomyosarcoma
The remaining subtypes of rhabdomyosarcoma include spindle cell/sclerosing rhabdomyosarcoma (SCR) and pleomorphic rhabdomyosarcoma (PR). Spindle cell/sclerosing rhabdomyosarcoma was separated from ERMS in the most recent 2013 WHO Blue Book [1]. Sclerosing rhabdomyosarcoma arises in the extremities across age groups. Spindle cell rhabdomyosarcomas arise in the paratestis of male children and the head and neck of adults (Fig. 13.10e). In pediatric cases, spindle cell rhabdomyosarcoma can provide a challenging differential with ERMS, with which it would have previously been classified. These cases generally show a good prognosis. However, in adult patients of spindle cell or sclerosing rhabdomyosarcoma, the prognosis is more guarded with higher rates of recurrence and metastasis [135]. Interestingly, very recent data suggest that a subset of spindle cell rhabdomyosarcomas in children are associated with rearrangements involving NCOA2, though this has not been observed in adult patients [136]. Further study of these rearrangements, which have involved fusions with SRF and TEAD1, including their prevalence, is anticipated, which may result in potential diagnostic utility of this observation.
PR is a high-grade, pleomorphic sarcoma arising essentially only among adults and showing varying degrees of skeletal muscle differentiation. These cases show pleomorphic, atypical rhabdomyoblasts (in contrast to the non-atypical rhabdomyoblasts of rhabdomyomas or the generally nonpleomorphic rhabdomyoblasts of ERMS and ARMS). The morphology is often similar to that of high-grade undifferentiated pleomorphic sarcoma (previously so-called “malignant fibrous histiocytoma”) but with varying degrees of skeletal muscle differentiation identifiable morphologically and by use of markers such as desmin and myogenin (MYOG also known as MYF4 see Fig. 13.10f). These are tumors of deep soft tissue of the extremities, often rapidly enlarging, with aggressive course. From a molecular standpoint they demonstrate complex karyotypes with multiple structural genomic abnormalities and none of the features of ARMS or ERMS, though generally they do not pose a diagnostic challenge for which testing for the recurrent PAX3/7–FOXO1 abnormalities of ARMS would be useful.
Vascular Tumors of Soft Tissue
Vascular tumors recognized by the WHO include a wide morphologic array of tumors, ranging from benign hemangiomas to intermediate (locally aggressive/rarely metastatic) hemangioendotherliomas and fully malignant sarcomas, which include both angiosarcoma of soft tissue and epithelioid hemangioendothelioma [1]. This is an area where actionable data regarding the molecular pathogenesis of these tumors has only begun to emerge; in fact, really only the latter two malignant vascular tumors have molecular tests in broad clinical use at this time. It may be anticipated that that in coming years, with accelerating use of Next-Generation molecular technologies, that additional, testable molecular lesions will be identified in the benign and intermediate lesions in this class. The first two examples described herein, that of pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma, intermediate lesion) and epithelioid hemangioendothelioma (malignant) demonstrate the ability to harness Next-Generation Sequencing to identify potentially testable molecular lesions.
Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma (EHE) is a rare vascular malignancy composed of epithelioid cells showing endothelial differentiation with plump epithelioid morphology arrayed in a distinctive myxocollagenous stroma (Fig. 13.11). The cells are variably plump and spindled to stellate, with eosinophilic cytoplasm, and show remarkable propensity for formation of intracytoplasmic lumina that appear as intracytoplasmic vacuoles. These lesions arise across a broad age range, usually in adults, and with slight preference for females. EHE presents as a soft tissue or visceral mass, usually painful, and in half of cases associated with a pre-existing vessel. Behavior is difficult to predict for this lesion, and the metastatic rate, ~25 %, and disease-specific mortality ~15 %, has been associated with larger lesions and increased mitotic rate [137, 138].
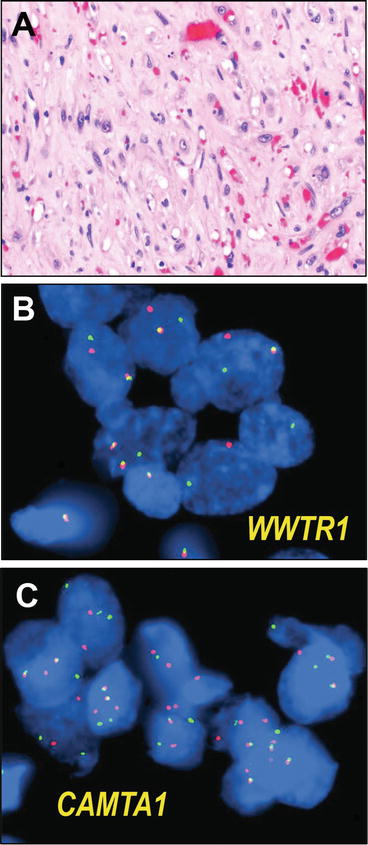
Fig. 13.11
Epithelioid hemangioendothelioma. Epithelioid hemangioendothelioma is a malignant vascular tumor composed of plump epithelioid to polygonal cells, growing in clusters, cords, and single cells, variably forming abortive vascular structures, including distinctive intracytoplasmic lumina. The intervening stroma is variably collagenous, to myxocollagenous or myxochondroid in appearance (a). Recent scholarship has identified a pathognomonic rearrangement, a WWTR1-CAMTA1 gene fusion in these tumors, which may be identified by break apart FISH, including assays using red and green flanking fluorescent probes for the WWTR1 locus (b) or the CAMTA1 locus (c). FISH micrographs courtesy of Dr. Brian Rubin and Dr. André Oliveira
The morphologic differential diagnosis for EHE is relatively broad, encompassing a spectrum of benign to malignant myxoid and vascular lesions with myxoid or chondroid stroma, particularly epithelioid hemangioma, chondroid lipoma, myxoid liposarcoma, myoepithelioma, myxoid chondrosarcoma, or even chordoma. The recent discovery, however, of a pathognomonic translocation t(1;3), fusing the genes WWTR1 and CAMTA1, resulting in overexpression of the c-terminal end of CAMTA1 under transcriptional regulation of WWTR in the majority of cases [139]. Furthermore, breakpoint analysis has shown that in multifocal EHE in the liver, the foci are monoclonal and likely represent locoregional metastases rather than independent primary tumors [140]. An additional gene fusion, that of YAP1 and TFE3, has been seen in smaller subset of EHE occurring in younger patients and showing more vasoformative features [141], rendering this diagnostically challenging lesion imminently testable by routine fluorescence in situ hybridization (Fig. 13.11) or RT-PCR-based methodologies.
Pseudomyogenic Hemangioendothelioma
Similarly, pseudomyogenic hemangioendothelioma (PH), also described as epithelioid sarcoma-like hemangioendothelioma [142], is an infrequent but distinctive vascular tumor of intermediate biological potential (rarely metastasizing) that various authors have more likened, in terms of morphology, to epithelioid sarcoma [142] (see below) or a myogenic tumor [143]. It most frequently involves the lower extremities of young adults. Descriptions have varied, though the current WHO Blue Book describes a cellular neoplasm with spindled morphology with plump eosinophilic cytoplasm (Fig. 13.12) that may simulate rhabdomyoblastic differentiation [1], though this lesion is thought to encompass many tumors described previously as the “fibroma-like” variant of epithelioid sarcoma [142, 143]. Rare intracytoplasmic lumina have been described, though true vascular formation has not been noted. A distinct propensity to recur has been noted in over half of patients, though metastasis is infrequent [143].
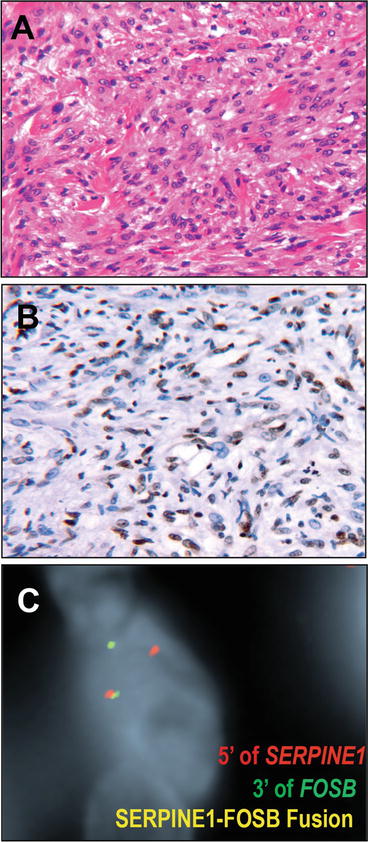
Fig. 13.12
Pseudomyogenic hemangioendothelioma. Pseudomyogenic hemangioendothelioma (epithelioid sarcoma-like hemangioendothelioma) is an intermediate (rarely metastasizing) vascular neoplasm characterized by fascicles to sheets of cells with vesicular nuclei and polygonal to spindled morphology with striking eosinophilic cytoplasm (a). Some have likened the morphology to fibroma-like epithelioid sarcoma, while to others this is more evocative of rhabdomyoblastic morphology. Supportive of endothelial differentiation, immunohistochemistry for vascular markers ERG (b) and CD31 are diffusely positive (b) as are keratins. In contrast, CD34 is negative. Recent data have identified a characteristic SERPINE1-FOSB fusion in these tumors, illustrated by this dual color single fusion FISH fluorescence micrograph using a red probe upstream of SERPINE1 and green probe downstream of FOSB, resulting in a yellow fusion signal if rearranged. FISH micrographs courtesy of Dr. Charles Walther and Dr. Fredrik Mertens
Very recent reports identify a testable, pathognomonic molecular lesion in PH. Proceeding form observation of recurrent t(7;19) translocation in PH [144], Walther et al. identified a novel, recurrent fusion of the genes SERPINE1 and FOSB in PH by a combination of cytogenetics, RNA seq, and RT-PCR [145]. This fusion is thought to result in coupling expression of FOSB transcription factor (a member of the FOS family of proteins that dimerize with JUN family transcription factors to form the activating protein 1 AP-1 complex) to the constitutively strong expression of SERPINE1, in vascular cells. While this fusion may be detected by a variety of techniques, it is certainly testable by break apart or dual color fusion FISH methods (Fig. 13.12).
Angiosarcoma
Angiosarcoma of soft tissue is a malignant sarcoma showing endothelial differentiation. These tumors are highly malignant and arise of a broad age range, though with most cases arising in older adults, most often males. Subsets of cases, particularly cutaneous examples, are associated with prior exposures, including radiation [146, 147], lymphedema (Stewart-Treves Syndrome) [6, 148, 149], or foreign bodies, including vascular grafts [150, 151] though angiosarcomas of deep soft tissues occur most commonly in the extremities, head and neck, and retroperitoneum, mediastinum, and mesentery [1, 152, 153]. Histologically these tumors are characterized by haphazard and infiltrative anastomosing vascular spaces compose of endothelial cells with varying degrees of atypia (Fig. 13.13). Tufting, stratification, and even formation of papillae of the usually mitotically active neoplastic endothelium is characteristic, though some areas may be more solid, and the malignant vascular spaces are not surrounded, at least not completely, by perivascular myoid cells. Epithelioid cases closely simulating carcinoma have been described and present recurrent diagnostic challenge [154]. These sarcomas show a distinctly poor prognosis, with a median survival less than 1 year after diagnosis [152, 153].
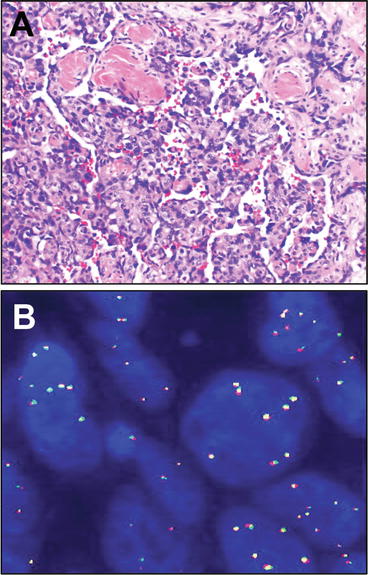
Fig. 13.13
Angiosaroma of soft tissues. Angiosarcoma of soft tissues is a malignant sarcoma showing endothelial differentiation. These tumors range from well-differentiated tumors that may be difficult to separate morphologically in biopsy samples from atypical vascular lesions such as arise (similar to angiosarcoma) in the post-radiation setting, to tumors with overtly malignant, even epithelioid morphology. The morphologic hallmark across this spectrum, is proliferation of infiltrative, irregularly anastomosing vascular lumina lined by atypical, hyperchromatic endothelial cells (a), often showing stratification or even papillation. Secondary angiosarcomas such as arise in the setting of chronic lymphedema, or increasingly, in the post radiation setting, demonstrate amplification of the MYC (c-myc) locus, illustrated here by multiple signals for the MYC locus. In this example, break apart red and green fluorescent probes flanking the locus do not show rearrangement but rather show low level amplification present as a number of copies per cell (b). FISH micrograph courtesy of Dr. Colleen Jackson-Cook
The setting where molecular testing has found use in angiosarcoma concerns the increasing number of vascular lesions seen in the post-radiotherapy setting of an aging population, and in particular that of post-treatment for breast cancer, where radiotherapy has become a mainstay of organ preserving therapy [147, 155, 156]. Not only do post-radiation angiosarcomas arise in this setting, but also a class of atypical vascular lesions (AVLs) [157], which may strongly simulate well-differentiated cutaneous angiosarcoma yet do not show the aggressive course that is so characteristic of angiosarcomas [158, 159]. Thus, the observation, relatively recently [160], of very prevalent amplification of the proto-oncogenic transcription factor, MYC, in post-radiation and lymphedema-associated angiosarcomas [160, 161] has proven to be quite useful as a diagnostic marker by FISH [162] (Fig. 13.13). Co-amplification of FLT4 is seen in about a quarter of cases [163]. Notably, similar to other tumors where genomic rearrangements lead to overexpression, MYC overexpression may be detected by immunohistochemistry, though further study will be necessary to assess the sensitivity and specificity of MYC immunohistochemistry to MYC amplification and to post-radiation angiosarcoma as opposed to other lesions in the differential [164, 165]. Finally, emerging data implicate prevalent MYC amplification (by FISH) in post-radiation sarcomas of other histologies [166], a feature with implications not only for use of diagnostic molecular testing but also development of potential therapeutic strategies. It should be noted that a subset of primary angiosarcomas are also characterized by MYC amplification, and thus its observation should not be construed as specific to secondary (lymphedema or radiation-related) angiosarcoma [167].
Gastrointestinal Stromal Tumors
Gastrointestinal stromal tumors (GISTs) are the most common primary mesenchymal neoplasm of the gastrointestinal tract, and show variable morphology and biological potential. These tumors were originally thought to represent smooth muscle or even nerve sheath neoplasms as reviewed recently by Appelman [168] but subsequently appreciated to represent neoplasms derived from the interstitial cells of Cajal, or their precursors, of the GI tract [22, 169–171]. These tumors arise in patients of any age (though pediatric examples often represent a somewhat distinct entity—see below) and without preference of male or female patients. They show variable prevalence by anatomic distribution, with approximately 60 % arising in the stomach, 25 % arising in the jejunum and ileum, and the remaining 15 % in the duodenum, esophagus, colon, and rectum.
From the standpoint of histopathology, these tumors may show a remarkable range of appearances, with tumors showing variable spindle cell and epithelioid cytomorphology, generally a lobular architecture, and usually a lack of nuclear pleomorphism, excepting exceptional cases showing “dedifferentiated” or “sarcomatoid” morphology [172]. More variable features include perinuclear cytoplasmic vacuolation, nuclear palisading (simulating the Verocay bodies of schwannomas) (Fig. 13.14) and multinucleated cells. These cell types are arrayed in a stroma that may be myxoid or even chondromyxoid, usually without inflammation. Notable in some examples are “skeinoid fibers,” which are distinctive extracellular collagen deposits more characteristic of GISTs of the small and large bowel than gastric examples. While these tumors frequently erode the mucosa with luminal bleeding, only a subset show invasion of the mucosa and tumor cell necrosis, both of which are negative prognostic factors. Overall GISTs of the stomach are less aggressive than those of the small bowel; the most important prognostic factors thus are anatomic site, size, and mitotic index, which have been implemented into useful nomograms stratifying GISTs by risk of recurrence or metastasis based on the cumulative work of the Armed Forces Institute of Pathology [170]. These tumors may spread by either hematogenous dissemination, most commonly to the liver, but also to bones and soft tissues, or spread through transperitoneal seeding. The differential diagnosis of GISTs, which includes a number of lesions such as schwannoma, leiomyoma, inflammatory fibroid polyp, and fibromatosis, had been difficulty in the past, though contemporary immunohistochemical markers, including KIT (Fig. 13.14) and DOG-1 (ANO1) show a high degree of utility as sensitive and specific markers for this tumor [22, 171, 173].
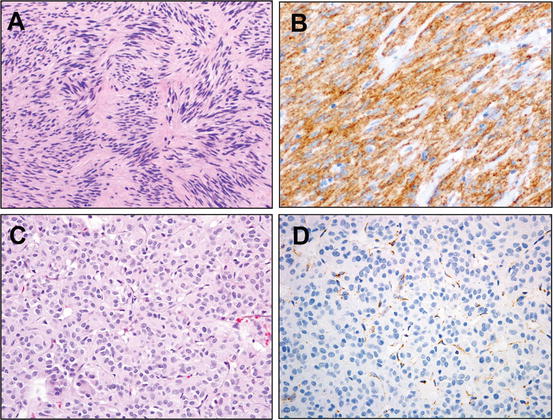
Fig. 13.14




Gastrointestinal stromal tumors. Gastrointestinal stromal tumors (GISTs) demonstrate variable morphology, ranging from spindle cells (an example with nuclear palisading pictured, a) to epithelioid and sarcomatoid. Though originally thought to be smooth muscle tumors, GISTs are now known to be neoplasms of the interstitial cells of Cajal, with which they share immunohistochemical positivity, in >90 % of cases, for the membrane receptor tyrosine kinase KIT (b). Mutations of KIT are detected in ~75 % of GISTs, while approximately 10 % show mutation of the related receptor tyrosine kinase PDGFRA. Of the remaining GISTs, wild type for KIT and PDGFRA, emerging data relate a subset of cases to mutation or, at least, loss of expression of the tetrameric succinate dehydrogenase complex subunits SDHA, SDHB, SDHC, or SDHD. Such cases often show prominent epithelioid morphology (c), while SDHB immunohistochemistry, where expression is lost irrespective of which SDH subunit is anomalous but maintained in vasculature (d), has been proposed as a robust means to interrogate the status of this pathway in wild-type GISTs
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
