Ewing sarcoma
t(11;22)(q24;q12)
EWS-FLI1
t(21;22)(q22;q12)
EWS-ERG
EWS-AFT1
Clear-cell sarcoma
t(12;22)(q13;q12)
EWS-WT1
Desmoplastic small round cell tumor
t(11;22)(q13;q12)
EWS-CHN
Extraskeletal myxoid chondrosarcoma
t(9;22)(q22;q12)
TAF2N-CHN
TLS-CHOP
t(9;17)(q22;q11)
TLS-ATF1
PAX3-FKHR
Myxoid liposarcoma
t(12;16)(q13;p13)
PAX7-FKHR
Angiomatoid fibrous histiocytoma
t(12;16)(q13;p11)
SYT-SSX1,2
Alveolar rhabdomyosarcoma
t(2;13)(q35;q14)
t(1;13)(p36;q14)
COL1A1-PDGFB
ETV6-NTKR3
STS have been notoriously difficult to account for accurately in cancer registries. Traditionally registry classification schemes are categorized by organ site, as adult tumors are predominantly epithelial malignancies of each organ. Soft tissue sarcomas then are only counted when they occurred in a connective tissue (vessel, nerve, muscle, fibrous tissue). This likely underestimates the number of STS, which can occur within or in close proximity to an organ. Alternatively, the International Childhood Cancer Classification pediatric classification scheme, which is used for the AYA site recode in the Surveillance, Epidemiology, and End Results (SEER) registry, recognizes and divides STS by histology but reflects the predominance of rhabdomyosarcoma (RMS) in children by only having three other subgroups: fibromatous (fibrosarcoma, peripheral nerve, and other fibrous), Kaposi sarcoma, and other STS (specified and unspecified).
In 2013, the World Health Organization (WHO) classification of STS was rewritten for the first time in 11 years [8]. The new inclusion of malignant peripheral nerve sheath tumor (MPNST), dermatofibrosarcoma protuberans (DFSP), and gastrointestinal stromal tumor (GIST) in STS classification rather than nervous system, skin and gastro-intestinal tract classification respectively, is particularly important in the AYA age range where these tumor types are quite common. Table 15.2 outlines the incidence and classification of the most common AYA soft tissue sarcomas by WHO classification and AYA site recode. Note that neither the SEER nor the WHO classification includes phyllodes tumor of the breast, mesothelioma, or any of the non-leiomyosarcoma STS of the uterus (endometrial stromal tumor, mixed Mullerian tumor, etc.), and they are not included in the figures or discussion. The natural history and treatment of extraosseous Ewing sarcoma, osteosarcoma, and chondrosarcoma are comparable to their more common presentation in the bone and are discussed in Chap. 12. Lastly, Kaposi sarcoma is included in SEER and WHO classifications but is a distinct STS in epidemiology, etiology, treatment, and outcome and dramatically skews calculations and conclusions and, unless otherwise noted in the text or figures, is not included in reference to “soft tissue sarcoma (STS).”
Table 15.2
Subtypes of soft tissue sarcoma
Subtype | ICD-O-3 | ICCC and AYA subgroup | WHO subgroup | Rate in AYAs (per million) | Percent of total STS in AYAS |
|---|---|---|---|---|---|
Kaposi sarcoma | 9140 | Other, Kaposi | Vascular | 5.6 | 19.8 |
Dermatofibrosarcoma protuberans | 8832–3 | Fibromatous | Fibroblastic | 5.0 | 17.7 |
Liposarcoma | 8850–5, 8857–8, 8860 | Other, specified | Adipocytic | 2.3 | 8.1 |
Sarcoma NOS/undifferentiated/spindle cell/giant cell/small cell (non-bone site) | 8800–3, 8805 | Other, unspecified | Tumor of uncertain differentiation | 2.3 | 8.1 |
Leiomyosarcoma | 8890–1, 8893–6 | Other, specified | Smooth muscle | 2.3 | 8.0 |
Synovial sarcoma | 9040–3 | Other, specified | Tumor of uncertain differentiation | 2.1 | 7.2 |
Rhabdomyosarcoma (RMS) | 8900–2, 8910, 8912, 8920 | RMS | Skeletal muscle | 1.5 | 5.1 |
Malignant peripheral nerve sheath tumor (non-CNS site) | 9540, 9560–1, 9571 | Other, specified | Nerve sheath | 1.3 | 4.7 |
Gastrointestinal stromal tumor | 8936 | – | GIST | 1.1 | 3.8 |
Fibromatous/fibroblastic | 8811, 8821–24 | Fibromatous | Fibroblastic | 1.1 | 3.8 |
8825 | Other, specified | ||||
Fibrohistiocytic | 8830, 8835, 9252 | Fibromatous | Fibrohistiocytic | 1.0 | 3.5 |
9251 | Other, specified | Fibrohistiocytic | |||
8836 | Fibromatous | Tumor of uncertain differentiation | |||
Hemangiosarcoma/vascular STS | 9120, 9125, 9130, 9133, 9170 | Other, specified | Vascular | 0.8 | 2.9 |
Epithelioid | 8804 | Other, specified | Tumor of uncertain differentiation | 0.5 | 1.8 |
Other: | 8991 | RMS | Tumor of uncertain differentiation | 0.5 | 1.8 |
Embryonal sarcoma | 8840 | Other, specified | |||
Myxosarcoma | 8842 | ||||
Ossifying fibromyxoid tumor | 8982 | ||||
Malignant myoepithelioma | 8990 | ||||
Malignant mesenchymoma | 9044 | ||||
Clear-cell sarcoma, not of kidney | 9580 | ||||
Malignant Granular cell tumor | |||||
DSRCT | 8806 | Other, unspecified | Tumor of uncertain differentiation | 0.5 | 1.6 |
Solitary fibrous tumor/hemangiopericytoma | 8815 | Fibromatous | Fibroblastic | 0.3 | 1.0 |
9150 | Other, specified | ||||
Alveolar soft-part sarcoma | 9581 | Other, specified | Tumor of uncertain differentiation | 0.3 | 1.0 |
Total | 28.5 | 100 % |
STS may be classified also according to their grade of malignancy. Tumor grade is determined by a combined assessment of histological features: degree of cellularity, cellular pleomorphism or anaplasia, mitotic activity, and degree of necrosis. Different histotypes with the same grade of malignancy could display the same clinical behavior. In general, low-grade tumors may have local aggressiveness but a low tendency to metastatic spread. High-grade tumors have a more invasive behavior with a high propensity to metastasize (in particular to the lung). Some histotypes (i.e., rhabdomyosarcoma, but also synovial sarcoma, alveolar soft-part sarcoma, and angiosarcoma) are usually considered as being high grade independently from their mitotic index, necrosis, and cellularity.
Different grading systems (generally three-grade systems) have been defined over the years by pediatric and adult oncologists for predicting clinical course and prognosis and defining a risk-adapted treatment. The most frequently used grading systems for adult sarcomas are the National Cancer Institute (NCI) system and the French Federation of Cancer Centers (FNCLCC) system [9]. The Pediatric Oncology Group system is similar to the NCI system but accounts for tumors found exclusively in childhood [10].
15.3 Epidemiology
Overall, STS are rare: with an annual incidence of around 5/100,000 persons of all ages, they account for <1 % of all cancer in the United States. Like many cancers, STS incidence increases exponentially with age. Rates in the AYA age range start at 1.4/100,000 for 15–19-year-olds and rise gradually to 3.4/100,000 in 35–39-year-olds. A total of approximately 2,900 STS are diagnosed in AYAs each year in the United States.
In contrast to this rising incidence with increasing age, STS become a less common proportion of all cancers (Fig. 15.1). Whereas in children under 15, STS account for nearly 7 % of cancer, this percent drops to 5 % of cancer in the third decade and 3 % in the fourth decade. Across the AYA age spectrum, histotypes of STS change as well (Fig. 15.2). SEER data from the period 2000–2011 shows DFSP to be the most common non-Kaposi STS among 15- to 39-year-olds, followed by liposarcoma, sarcoma not-otherwise specified (NOS), leiomyosarcoma, fibromatous sarcoma, synovial sarcoma (SS), RMS, and MPNST.
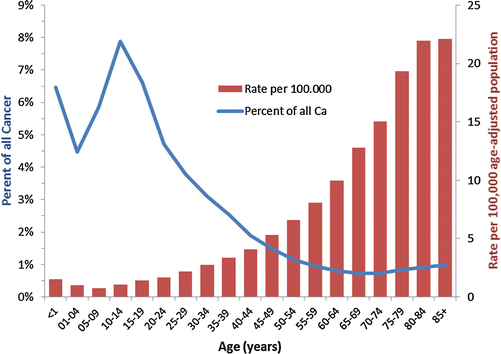
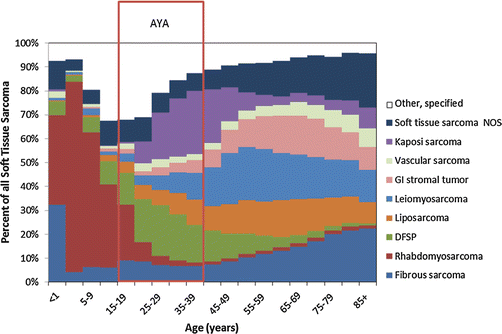
Fig. 15.1
Incidence of soft tissue sarcoma as a percent of all cancer (left axis) and rate per 100,000 and age-adjusted to the 2000 US standard population (right axis), by age, SEER 18, 2000–201
Fig. 15.2
Distribution of soft tissue sarcoma subtypes within 5-year age intervals from birth to age 85+, SEER 18, 2000–2011. NOS not otherwise specified, GI gastrointestinal, DFSP dermatofibrosarcoma protuberans
Between 2000 and 2011, the incidence of STS was nearly the same between females and males. However there are marked differences in incidence by race: for all categories (RMS, fibromatous, and other STS), AYA blacks have the highest rate, followed by non-Hispanic whites, then Hispanics, Asians, and American Indians/Natives (Fig. 15.3). The incidence of STS in AYAs has not changed dramatically in the last 36 years; the one particular rise in incidence is the rising rate in AYA males of “other STS” (non-fibromatous and non-RMS), which has increased from 1.0/100,000 to 1.5/100,000 (Fig. 15.4).
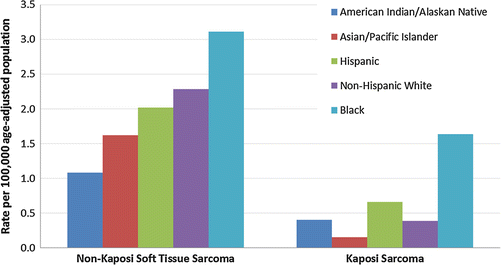
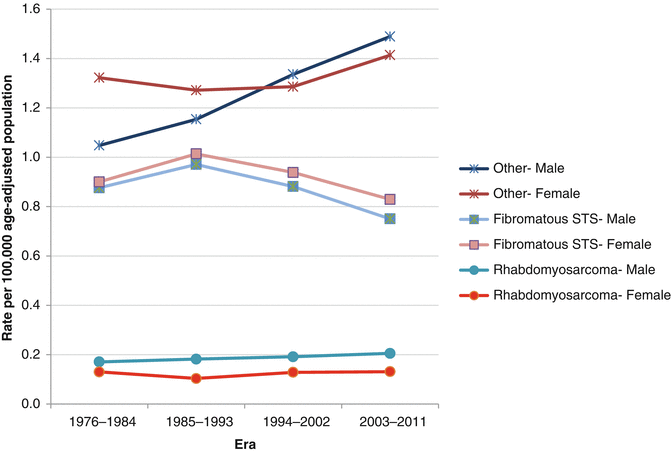
Fig. 15.3
Incidence of non-Kaposi soft tissue sarcoma (left) and Kaposi sarcoma (right) in AYAs age 15–39 according to race/ethnicity, SEER 18, 2000–2011
Fig. 15.4
Change in the incidence rate of soft tissue sarcoma in AYAs age 15–39, by sex and subtype, as a function of era of diagnosis, SEER 18, 1976–2011. Fibromatous STS = International Childhood Cancer Classification of fibromatous soft tissue sarcoma (STS) including dermatofibrosarcoma protuberans, solitary fibrous tumor, fibromatous, fibroblastic, and fibrohistiocytic sarcomas. Others= all non-Kaposi, non-fibromatous, and non-rhabdomyosarcoma STS
The epidemiology of Kaposi sarcoma bears particular note: it is still the most common STS in AYAs, although its incidence has dropped to nearly the incidence of the pre-AIDS epidemic of the 1990s. It is almost exclusively seen in males, and the rate in black AYAs is three times that in non-blacks (Fig. 15.3).
Concerning survival, in the SEER 2000–2011 registry, the 5-year overall survival (OS) rate for STS in AYAs averages 73 % with substantial differences according to the histology, the grade of malignancy, and the stage of the disease [3]. Compared to other ages, AYAs with RMS have worse survival than younger patients, and AYAs with Kaposi sarcoma have worse survival than older patients. A fascinating trend is seen in examining STS survival by sex. Specifically between the ages of 10 and 45, the overall 5-year survival is worse in males than females; in both younger and older ages, males have better survival (Fig. 15.5). Notably, for AYAs with STS, especially those with “other STS,” there has been no improvement in survival for the last 25 years. For AYAs age 15–39, 5-year cause-specific survival rates for STS in 2003–2010 were 76.2 %, little better than 75.0 % in 1976–1984, whereas survival for those <15 has improved from 71.7 to 76.6 % and for those over 40 has increased from 51.9 to 62.0 % (Fig. 15.6).
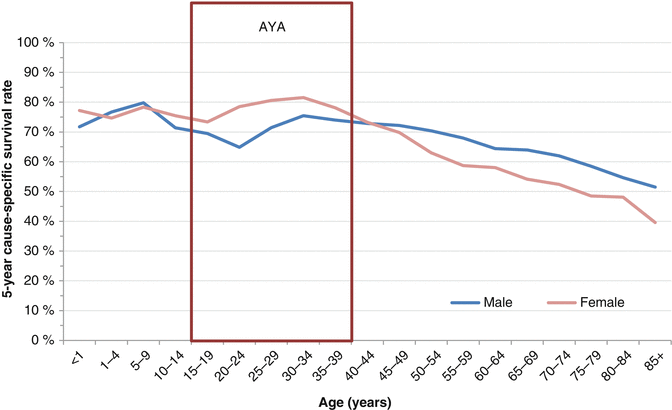
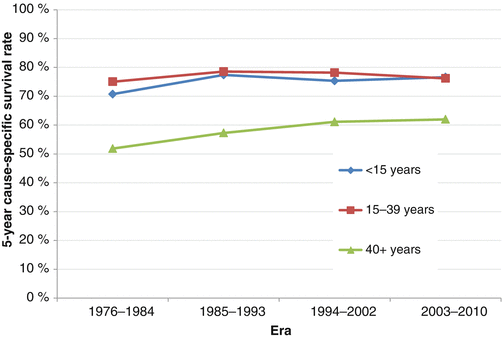
Fig. 15.5
A 5-year cause-specific survival of soft tissue sarcoma by sex, as a function of age, SEER 18, 2000–2011
Fig. 15.6
Change in 5-year cause-specific survival rates of soft tissue sarcoma for those aged <15, 15–39, and 40+ at diagnosis, as a function of era, SEER 18, 1976–2010
15.4 Etiology
For most STS subtypes, the pathogenesis remains unknown and there are few well-established risk factors [11]. Ionizing radiation clearly causes sarcomas, and chemical carcinogens (herbicides, polyvinyl chloride, and arsenic) have been associated with the development of some type of sarcomas, but the etiological relationship remains unclear. HHV8 is known to be the causative agent for Kaposi sarcoma. Although they cumulatively cause few of all STS, several genetic predispositions are well described. Neurofibromatosis type 1 increases the lifetime risk of MPNST to approximately 10 % and appears to increase the rate of RMS as well [12, 13, 97]. STS is one of the defining tumors of Li-Fraumeni syndrome, and survivors of hereditary retinoblastoma have a cumulative risk of developing a postradiation STS of 13.1 % [14]. Finally, there are several described germline mutations associated with GIST predisposition (particularly Carney-Stratakis syndrome) [15]. Those with genetic conditions are predisposed to develop tumors at a younger age, so that the proportion of adolescents and young adults with STS with a genetic predisposition is probably higher than in older adults [16].
15.5 Diagnosis
15.5.1 Clinical Presentation
The initial signs and symptoms in patients with STS may vary and depend on the site of origin and tumor extension, as well as on the different degree of malignancy along histotype and tumor grade. An enlarging painless mass is the most common presentation. In 15- to 29-year-olds, about one-third of STS originate in the extremities. For example, for SS, the clinical presentation is often a slow-growing mass in the soft tissues of the lower extremity, especially around the knee. However, STS can arise anywhere in the body. RMS may occur also in sites in which striated muscle tissue is normally absent: the head and neck region represents the most common location for RMS, and the symptoms vary from proptosis, cranial nerve palsy, or nasal obstruction; hematuria may be present in RMS of the genitourinary tract; ascites and intestinal obstruction can occur with retroperitoneal tumors.
For low-grade STS, growth rate may be indolent, and sometimes the diagnosis is done after removing a small swelling that has existed for several years. The clinical presentation is very different among the different entities included in this group of tumor: e.g., MPNST are generally axial (trunk, head-neck region) and aggressiveness disease; epithelioid sarcomas present typical features such as peculiar superficial distal location like hand and fingers, indolent growth, and tendency for lymph node involvement; desmoplastic small round cell tumors (DSRCT) usually present as rapidly growing and large abdominal masses, generally disseminated at the time of diagnosis, with peritoneal seeding.
15.5.2 Diagnostic Procedures
In the case of suspected lesions, three diagnostic levels need to be evaluated: (a) the histological diagnosis, (b) the definition of locoregional extension, and (c) the staging of the disease to detect regional or distant metastases.
Pathological assessment is necessary to define the histological diagnosis. The initial biopsy has the aim to define the diagnosis but also should provide enough material for immunochemistry, cytogenetics, biological studies, and central pathology review for patients to be included in multicentric clinical trials. In the case of a large and deep soft tissue mass, biopsy should be always the initial surgical procedure, in order to avoid inadequate surgery. Open biopsy (incisional biopsy) or core needle biopsy (Tru-Cut, guided by ultrasound or computed tomography [CT] scan) is preferred to fine-needle aspirates that could establish the presence of malignancy but rarely identify the subtype or provide the tissue required for additional studies. In any case, the initial biopsy should be carefully planned by an experienced surgeon, taking into account the possible subsequent definitive surgery that must include the scar and the biopsy tract. For example, in STS of the extremities, the incision must be longitudinal to the limb and not traverse multiple compartments; very careful hemostasis must be ensured to minimize the risk of postsurgical hematoma and need for drains.
Local extent of disease is determined by CT scan or magnetic resonance imaging (MRI), the latter appearing to be superior in defining soft tissue extension.
The diagnostic workup is completed by staging investigations, aimed to detect regional and distant metastases. Since distant metastases mainly occur to the lung, chest CT scan is the most important exam. Abdominal ultrasound or CT scan may be used to identify abdominal metastases, while technetium bone scan can be required to detect bone metastases (for patients with aggressive and high-grade STS) [17, 64, 72]. Positron emission tomography is increasingly used, though its role remains not well defined in STS [18].
15.5.3 Staging
An adequate stratification of the patients is necessary for a risk-adapted therapy. However, as in grading, pediatric and medical oncologists have not used the same systems, making comparison of risk and prognosis difficult. The pediatric Intergroup Rhabdomyosarcoma Study (IRS) postsurgical grouping system [19] supplements the pretreatment clinical tumor-node-metastases (TNM) classification [20], categorizing patients into four groups based on the amount and extent of residual tumor after the initial surgical procedure. Group I includes completely excised tumors with negative microscopic margins (also called R0 resection); group II indicates grossly resected tumors with microscopic residual disease (R1 resection) and/or regional lymph nodal spread; group III includes patients with gross residual disease after incomplete resection (R2 resection) or biopsy sampling; group IV encompasses patients with metastases at onset [19]. According to the TNM classification, T1 are those tumors confined to the organ or tissue of origin, while T2 lesions invade contiguous structures; T1 and T2 are further classified as A or B depending on whether tumor diameter is ≤ or >5 cm, respectively. Regional node involvement is defined as N0 or N1 and the status of distant metastases at onset as M0 or M1 [20].
However, adult oncology groups have generally utilized other systems: the Musculoskeletal Tumor Society Staging System requires the accurate definition of compartmentalization and the American Joint Committee on Cancer Staging System combines TNM definitions and histological grading [21].
15.6 Treatment Management and Outcome
The treatment of patients with STS is complex and necessarily multidisciplinary, requiring evaluation and management by a team with adequate expertise and experience and the ability to enroll on clinical trials (NCCN guidelines 1.2015). Treating patients outside of a referral center has been identified as an independent risk factor for nonadherence to treatment guidelines [22] which has in turn been seen to increase treatment cost, risk of recurrence, and mortality in STS [23].
15.6.1 Surgical Aspects
The approach to surgical resection of soft tissue sarcoma (STS) is focused on achieving an adequate resection margin. The goal of the orthopedic or surgical oncologist, regardless of the anatomic location of the tumor, is ideally to achieve a negative resection margin, which can be defined as grossly complete removal of the tumor with a surrounding rim of normal tissue, with no macroscopic tumor visible and no microscopic tumor cells present at the edge of the resected specimen. The ability to achieve a negative margin resection for STS, and the quality and quantity of the resected tissue surrounding the actual tumor mass, is largely determined by the tumor size and location, proximity of the tumor to critical structures that would lead to unwanted morbidity if removed en bloc with the tumor and the experience of the surgeon.
Any discussion of surgical margins requires an understanding of the local biology of STS as well as the concept of compartmentalization in the extremities as popularized by [24]. Sarcomas must be removed with a rim of surrounding normal tissue in order to achieve a negative resection margin. This is critical to avoiding local tumor recurrence because STS do not have a true capsule; instead as they grow, they compress the surrounding tissues into a reactive “pseudocapsule” that does not provide an actual barrier to tumor extension and actually contains malignant cells (Fig. 15.7). Beyond the “pseudocapsule” and surrounding the STS is a zone of variable distance that may look normal to the eye of the surgeon but which contains subclinical or microscopic disease and has been termed the “inflammatory” or “reactive” zone. If one could identify the extent of this zone and thereby the furthest possible extent of microscopic tumor cells beyond the main tumor mass, then placing the surgical margin beyond that point, through normal tissue, would always provide not just a negative resection margin but a locally curative one too, since no residual tumor cells would be left behind which could subsequently grow and account for a local tumor recurrence. There is currently no objective test which can identify the local extent of the microscopic disease within the “reactive” zone, although the presence of “peritumoral edema” that is often seen to surround STS on MRI scans may reflect areas at risk for harboring microscopic disease, that should either be resected at the time of surgery or be included in the radiotherapy field if treatment will involve combined management with preoperative radiation, although this is not a universally accepted practice [25, 26].
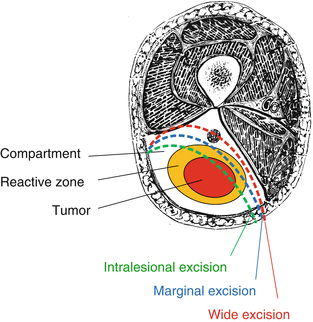
Fig. 15.7
Tumor, reactive zone and compartment, and type of resection, according to Enneking definition (The Authors thank Luna Boschetti for the picture)
Compartmentalization is based on fascial boundaries which generally act as impenetrable and predictable barriers to tumor growth. For instance, the superficial investing fascia in the extremities and the superficial fascia of the trunk or abdominal wall provide a barrier to deep tumor spread. Similarly, the superficial investing fascia in the extremities joins the deep intermuscular septae and defines muscular compartments which also act as barriers to local tumor spread. For STS that originate within a muscular compartment, tumors may grow longitudinally along fascial planes but typically cannot grow in a radial pattern beyond the fascia which acts as a barrier to local growth. However, there are a number of locations in the extremities where fascial compartments do not exist, most frequently at areas of flexor creases such as the axilla, antecubital fossa, inguinal canal, and popliteal fossa, and in these anatomic sites, it is often difficult to achieve a “wide” resection margin around an STS, such that adjuvant therapy is often required as part of the treatment plan.
The concept of fascia and compartments as barriers can be utilized to define different types of resection margins [24]. For instance, Enneking defined a “wide” resection margin as having either an intact fascial boundary (even if directly adjacent to or within a few millimeters of the tumor) or an adequate surrounding layer of tissues which would indicate that the tumor was removed with an intact pseudocapsule, reactive zone, and some additional normal tissue. Defining a fascial boundary is relatively straightforward; however, defining where the normal layer is situated beyond the reactive zone surrounding a STS is more challenging. As a result, local management of extremity STS historically necessitated entire compartment resections, and even more radical resections including amputations were commonplace to avoid local tumor recurrences.
With better local imaging by MRI which allows improved definition of local tumor extent, as well as the introduction of radiation as a commonly utilized treatment modality as part of combined management for the local control of STS, more conservative and function preserving surgery for STS is now possible. Generally, a 1–2 cm layer of normal tissue surrounding an STS (e.g., 1 cm for low-grade and 2 cm for high-grade tumors) is considered to represent an adequate wide margin and should provide local control in >90 % of cases following surgical resection alone [27, 28]. However, the majority of STS cannot be resected with a “wide” margin and therefore require combined management with (neo)adjuvant radiation or (neo)adjuvant chemotherapy or both. For extremity STS that require combined surgery and radiotherapy due to less than wide resection margins, a randomized clinical trial comparing preoperative and postoperative radiation by the Canadian Sarcoma Group showed that although preoperative radiation was associated with a higher risk of early postoperative wound healing complications, it was also associated with better long-term functional outcomes for patients due to less soft tissue fibrosis, pain, stiffness, and limb edema as well as less radiation-associated fractures [29, 30]. In that study, the rate of local and systemic control was identical in both treatment arms. A more recent phase 2 study of Image-Guided Intensity Modulated Radiation Therapy (IMRT) by the Toronto Sarcoma Group showed that this newer modality for delivery of preoperative radiation was safe and could diminish the risk of wound complications compared to conventional preoperative radiation yet maintain a low risk of local tumor recurrence as well as good limb function [31]. The use of preoperative radiation is becoming more commonplace and is likely the optimal approach for combined management of extremity STS as well as other difficult anatomic sites for most patients and is particularly useful in children and adolescents as it limits both the dose of radiation and the treatment volume [32].
Today, surgical resection margins are typically defined as R0 (negative margin), R1 (microscopically positive margin), and R2 (grossly positive margin). Gross positive (R2) margins are associated with unacceptably high rates of local recurrence. R0 does not differentiate between a true wide resection as defined by [24] where surgery alone is adequate, and a close but negative margin where the addition of radiation has been well documented to decrease the risk of local tumor relapse [33, 34]. Even in the setting of a microscopically positive margin (R1), the risk of local tumor relapse is significantly lower if adjuvant radiation is combined with surgery. A common anatomic scenario that can result in an R1 margin is when an STS is situated directly abutting a major motor nerve, blood vessel, or bone where resecting one of these structures would introduce significant morbidity for the patient. In this setting, planning for a very close margin of resection, such as using epineurium, vessel adventitia, or periosteum, in combination with preoperative radiation in order to save a critical structure has been shown to be safe [35, 36] even if the final margin ends up microscopically positive. The same studies also defined other situations in which resection of STS with positive margins (e.g., following a prior unplanned excision elsewhere or an unexpected positive margin during primary resection) leads to high rates of local recurrence and metastasis and therefore much worse outcomes for patients. Although much of this data is derived from the management of extremity STS, it likely applies to management of STS of other anatomic sites, including the head and neck, thorax, retroperitoneum and pelvis. For management of patients with retroperitoneal STS in particular, a consensus statement has recently been published by a transatlantic working group [37] that leads to initiation of a randomized clinical trial comparing surgery with or without preoperative radiation for patients with previously untreated nonmetastatic disease who are 18 years of age and over (EORTC 62092-22092 – STRASS clinical trial).
The pediatric approach to the management of STS has been quite different and focused on the role of chemotherapy, largely because of the chemosensitivity of rhabdomyosarcoma, soft tissue Ewing sarcoma/PNET, and possibly synovial sarcoma. In addition in pediatric patients, there is the added objective of trying to minimize the use of radiation in this young population. High-dose radiation in the extremity of a growing child can be complicated early on by growth arrest related to radiation of the growth plate, joint stiffness related to fibrosis, and bone osteopenia and weakness with subsequent risk of fracture. More importantly, and in the longer term, there is a time-dependent cumulative risk for development of secondary radiation-induced malignancies including sarcomas and even carcinomas, as well as other chronic health disorders depending on the location of the radiation including obesity, pulmonary, thyroid, and cardiac dysfunction [38]. As a result, most pediatric patients with any type of high-risk STS (i.e., large and high-grade tumors) tend to be treated with chemotherapy, even if they have an “adult” histologic type of tumor, in the hope that it will facilitate resection and minimize the need for radiotherapy. However, in the setting of inadequate surgical resection margins, radiation should still be utilized to enhance local tumor control.
Synovial sarcoma spans both the pediatric and adult age groups, and owing to the pediatric approach, most adults with high-risk synovial sarcoma have been offered preoperative chemotherapy over the past decade as part of their treatment (Fig. 15.8).
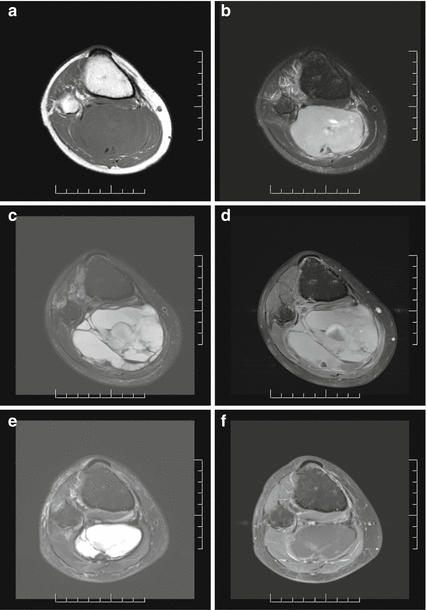
Fig. 15.8
A 28-year-old male with synovial sarcoma in the proximal calf/popliteal fossa. At diagnosis, (a) axial T1-weighted MRI image and (b) axial T2-weighted image with fat suppression show a large solid STS involving the superficial and deep posterior calf compartments and extending into the popliteal fossa with involvement of the neurovascular bundle. Following preoperative chemotherapy, (c) axial T2 with fat suppression and (d) axial T1 with fat suppression and gadolinium MRI images demonstrate interval enlargement and development of multiloculated cystic components with fluid levels, suggestive of hemorrhage and necrosis. Some rims of enhancement are seen on the post-gadolinium images. The patient received preoperative radiation following initial chemotherapy. (e) Axial T2 with fat suppression and (f) axial T1 with fat suppression and gadolinium MRI images show interval reduction in size of the lesion with less peripheral enhancement from gadolinium. Limb sparing surgery necessitated excision of the posterior tibia nerve as well as resection and reconstruction of the posterior tibia vessels. The final pathology report revealed 95 % treatment-induced necrosis. The patient remains alive without evidence of disease 2 years following treatment
15.6.2 Rhabdomyosarcoma
RMS is a distinct entity and clearly differs from other soft tissue sarcomas in regard to its natural history and its sensitivity to therapy: (a) it is one of the typical embryonal tumor of childhood, composed by cells resembling normal fetal skeletal muscle; (b) it is always characterized by high-grade malignancy, local invasiveness, and a marked propensity to metastasize, to the point that all RMS patients should be assumed to have micrometastatic disease at diagnosis and therefore need to be treated with systemic therapy; (c) it is generally characterized by good response to chemotherapy (80–90 % response rate) and radiotherapy.
RMS cells can be recognized by the expression of myosin and MyoD protein family antigen. Classically, two histological subtypes of RMS have been distinguished, the favorable embryonal subtype and the unfavorable alveolar subtype [39]. A third form, pleomorphic RMS, needs to be considered separately from other RMS subtypes (it is very rare in the pediatric population, occurring typically at an age older than 50 years, and it is probably more close to an adult sarcoma such as malignant fibrous histiocytoma or high-grade spindle-cell sarcomas than to other RMS subtypes typical of children). Cytogenetic and molecular analyses are of critical importance to define the diagnosis of RMS subtype. Most alveolar RMS (80–85 %) display a consistent genetic abnormality, that is, the reciprocal chromosomal translocations t(2;13) (q35;q14) or t(1;13)(p36;q14), generating the PAX3 or PAX7–FOXO1 chimeric protein. Embryonal RMS lacks a tumor-specific translocation but generally exhibits recurring abnormalities such as loss of heterozygosity on the short arm of chromosome 11 (11p15.5), which may act by inactivating tumor-suppression genes. A pattern of association between histotypes and clinical features has been described (i.e., the alveolar histotypes are more frequently localized at the extremities and in the trunk, and it is more typical of adolescents and young adults than of children). Recently, some controversies have emerged regarding the histopathological diagnosis of alveolar RMS: patients with alveolar histology but lacking the fusion gene PAX3 or PAX7–FOXO1(fusion gene-negative alveolar RMS) would have a genomic profile and a clinical course more similar to those of embryonal RMS than those of fusion-positive alveolar cases [40, 41]. These findings would suggests that a biological characterization would be better than the currently used histological assessment in classifying RMS subtype and predicting prognosis, and they will be probably incorporated in future risk stratification.
During the past 30 years, the 5-year overall survival (OS) rates of pediatric RMS have improved dramatically from 25 to 30 % to approximately 70 % [42–44, 46, 47, 51]. These results are due largely to the development of treatment approaches that are (a) multidisciplinary (including surgery, radiotherapy, and in particular multiagent effective chemotherapy), (b) risk-adapted (prognostic factors are used to stratify treatment: more intensive therapy improves cure rates in those patients with less favorable disease, whereas those with more favorable findings avoid overtreatment and side effects without jeopardizing survival), and (c) cooperative multi-institutional trials able to enroll a large number of patients. Historically, these trials included subjects up to the age of 18 or 21 years; however, in recent years, pediatric collaborative groups have raised the upper age limit of their RMS protocols to 30 years and more.
Currently, the two main cooperative groups dedicated to RMS are the Soft Tissue Sarcoma Committee of the North-American Children’s Oncology Group (COG) and the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG), which involves most of the European countries and others (e.g., Argentina, Brazil, Israel). Other European countries join the German CWS (Co-operative Weichteilsarkomen Studie) group.
The treatment approach of the different cooperative groups is built along similar lines, but differences still exist. RMS is a very heterogeneous disease, and the prognosis depends on multiple factors, including histological subtype, primary tumor site and size, lymph node involvement, and distant metastasis. For example, patients with alveolar histology continue to have less than optimal outcome, and most patients with distant metastasis do not achieve long-term cure. With the recognition of the different prognostic factors, the risk assignments (to decide the treatment’s intensity) became more complex but also more careful. The recent EpSSG protocol identifies low-, standard-, high-, and very high-risk groups (with 8 subgroups) for localized patients, plus the group of metastatic RMS cases [48, 65] (Fig. 15.9); the COG protocol describes three groups (low, intermediate, and high risk) and 17 subgroups, but the definitions do not cover the same subsets of patients (e.g., the EpSSG high-risk group grossly corresponds to the IRS-V intermediate-risk group) [49]. Among the other variables used in the risk stratification, the patient’s age has also emerged as a factor significantly influencing survival: in various series, patients over 10 years of age have been reported to have a worse prognosis than younger children [50].
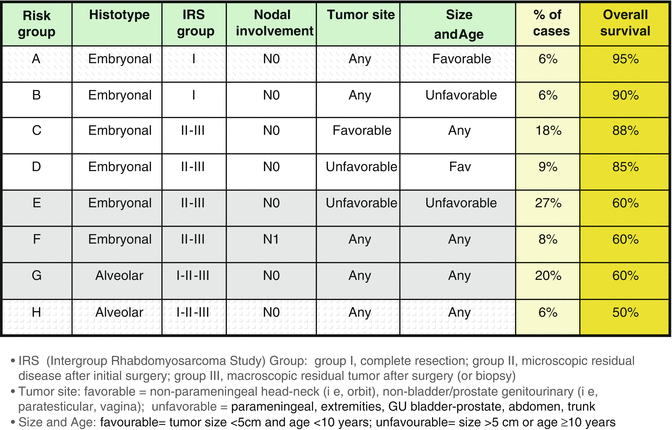
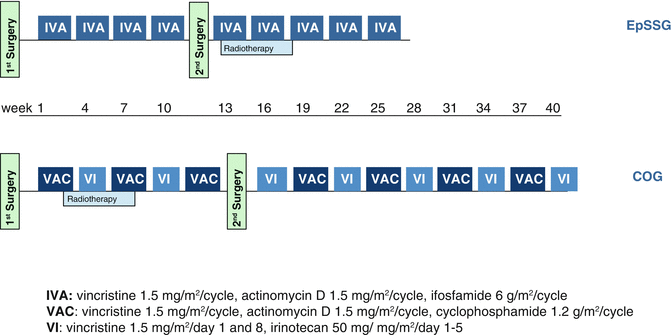
Fig. 15.9
Risk stratification, estimated percentage of patients, and estimated overall survival according to the European Pediatric Soft Tissue Sarcoma Study Group: (EpSSG) RMS 2005 protocol
Treatment strategy for RMS is necessarily based on a multimodal approach. An optimal local treatment is essential, because local progression or recurrence continues to be the primary causes of treatment failure. Nevertheless, it is possible to state that multiagent chemotherapy is the mainstay of the treatment and should be definitely given to all patients. The reliable chemosensitivity of RMS has led to an evolution in the role of local therapies: surgery has evolved over the years from being the primary treatment modality to just one component of a multidisciplinary approach and from an aggressive surgical approach to more conservative, organ-sparing procedures. As time of diagnosis, surgery with risk of anatomic or functional impairment is not recommended and biopsy should be performed. Tumors considered unresectable at diagnosis can be conservatively and completely resected in a large percentage of cases after tumor shrinkage achieved by primary chemotherapy. Similarly, the efficacy of chemotherapy may reduce the proportion of patients for whom radiotherapy is indicated (or the doses used) and thus reduce the risk of radiation-related sequelae, in particular for younger patients (fibrosis, bone and soft tissue hypoplasia, neuroendocrine dysfunctions, second tumors) [51]. Radiotherapy is often omitted in embryonal RMS after initial complete resection, but it is still recommended in many cases (i.e., alveolar RMS, inadequate resection, large tumors). Radiotherapy is generally delivered to the pretreatment tumor volume with doses generally ranging between 40 and 55 Gy, using three-dimensional conformal techniques.
The backbone of RMS treatment is intensive alkylating-based multidrug chemotherapy given for at least 6–9 months. A large number of different chemotherapeutic regimens have been tested over the years within cooperative randomized trials. In many protocols, the addition of various drugs to the gold standard VAC (combination of vincristine, actinomycin D, and cyclophosphamide, used in North America) and IVA regimen (which differs in the choice of the alkylating agent, i.e., ifosfamide in the place of cyclophosphamide, used in Europe) did not show clear advantage compared to the standard combinations.
In the recent EpSSG RMS 2005 trial, the role of doxorubicin has been evaluated in a randomized trial using the IVADO regimen (IVA plus doxorubicin) [52], administering early the maximum dose intensity of doxorubicin, compared to standard IVA. Preliminary results did not show any advantage for the addition of doxorubicin but a higher toxicity [53]. The trial remains open for a second randomization, to evaluate the role of maintenance therapy with low-dose continuous chemotherapy: patients with localized RMS who are in complete remission after 6-month chemotherapy are randomized to receive or not maintenance therapy with oral cyclophosphamide plus vinorelbine (which has shown activity in RMS) [54].
The most recent COG trials for localized RMS explored the role of camptothecin derivatives. In the COG D9803 study conducted between 1999 and 2005, the addition of topotecan to VAC failed to show benefit [46]. In the subsequent COG ARST0531 study conducted between 2006 and 2012, patients with localized standard-risk RMS have been randomized to receive 14 courses of VAC versus VAC alternating with vincristine-irinotecan (VI). As in other previous randomized trial adding new regimens to VAC, the addition of VI did not improve outcome; however, as there was minor hematologic/infective toxicity and a lower dose of alkylating agents, COG has decided that the VAC/VI regimen will be used in future trials as the comparator arm (Fig. 15.10) [55].