23 Skull-Base Tumors
Chemodectomas, Nonchromaffin Paragangliomas, Chordomas, and Chondrosarcomas*
Paragangliomas
Paragangliomas are benign neoplasms of neural crest origin that arise from small (0.1-0.5 mm) collections of neuroepithelial cells called paraganglia or glomus bodies.1 The paraganglion system is important during fetal development to supply catecholamines to the adrenal medulla as it matures and becomes functional.2 Most paraganglia subsequently regress, except along the autonomic nervous system and in certain organs. Paragangliomas are classified as chromaffin and nonchromaffin, depending on whether they produce catecholamines and thereby react with chromic acid.3 Adrenal paragangliomas are usually referred to as pheochromocytomas and are chromaffin-reaction positive, whereas tumors of the extra-adrenal paraganglionic tissue are not. Extra-adrenal paragangliomas of the head and neck are typically nonchromaffin and arise in the cervical region and temporal bone. Terms considered synonymous with paraganglioma include chemodectoma and glomus body tumor. Paragangliomas are named by their site of origin (e.g., carotid paraganglioma, jugular paraganglioma, tympanic paraganglioma, etc.), although the terms glomus-jugulare, glomus-tympanicum, glomus-vagale, and carotid-body tumors are still commonplace.4
Epidemiology and Genetics
Approximately 90% of all paragangliomas arise in the adrenal gland and are called pheochromocytomas. Most (85%) extra-adrenal paragangliomas arise in the abdomen. Approximately 3% occur in the head and neck.2
Paragangliomas of the head and neck are rare, accounting for 0.012% of all tumors5 and 1 out of every 30,000 head and neck tumors.6 Carotid-body tumors tend to be equally distributed between men and women, whereas temporal bone and vagale tumors occur more often in women.2,7–9 Some investigators suggest that higher altitudes and hypoxia may contribute to the development of carotid-body tumors.10,11 Paragangliomas generally occur between ages 50 and 60,7 but may occur earlier in patients with a family history. Between 1% and 3% have endocrine activity that causes symptoms similar to pheochromocytomas or carcinoid tumors.12 Although paragangliomas are normally benign, a small subset (10% or fewer) are malignant and metastasize.8,13 They are multifocal in 10% to 20% of cases, although multifocality can be as high as 33% to 55% in familial cases.9,14 Familial paragangliomas account for approximately 10% of all cases, and are much more likely to be multicentric and bilateral than sporadic tumors.3
Familial paragangliomas are inherited in an autosomal-dominant pattern, but display genomic imprinting with paternal inheritance, because the phenotype is expressed only when transmitted by the father.2,3,15–17 The responsible gene is PGL, which codes for SDH, a mitochondrial enzyme complex that plays an important role in oxidative phosphorylation and intracellular oxygen sensing and signaling.18 Within these complexes are specific subunits coded for using three distinct genes: SDHB, SDHC, and SDHD. Genetic analysis has identified three different genetic types of paraganglioma: paraganglioma 1 (PGL1) 11q23, paraganglioma 2 (PGL2) 11q13, and paraganglioma 3 (PGL3).19–22 PGL1 and PGL2 display the genetic imprinting pattern of inheritance, whereas PGL3 does not and is transmitted from either parent.19,22 Paragangliomas occur in syndromes containing multiple tumors, including multiple endocrine neoplasia type II,23 von Hippel-Lindau disease,24 neurogenic disorders such as von Recklinghausen neurofibromatosis type I,25 and Carney syndrome.26
Anatomy
The carotid body is composed of multiple chemoreceptors located at the bifurcation of the common carotid artery, which are responsible for detecting changes in the partial pressures of oxygen and carbon dioxide in arterial blood. It is the most common location for head and neck paragangliomas, comprising between 60% and 70% of cases.3 The temporal bone is the next most common location. Tumors arising along the superior jugular bulb are referred to as glomus-jugulare tumors, whereas those from the tympanic (Jacobson nerve) nerve of cranial nerve IX or the auricular (Arnold nerve) nerve of cranial nerve X are called glomus-tympanicum tumors. The former runs along the tympanic canaliculus from the inferior petrous portion of the temporal bone between the jugular fossa and the carotid canal to the floor of the tympanic cavity. The Arnold nerve (X) runs from the jugular fossa laterally into the mastoid process. Glomus-vagale tumors may arise along the vagus nerve in any of three vagal ganglia, although usually from the most inferior (nodose) ganglia. Other possible sites include the ciliary ganglion, nasal cavity, larynx, trachea, periaortic region, and fallopian tubes.27
Pathologic Conditions
Paraganglia are composed of two types of cells.2 Type I, also called chief or granular cells, are part of the amino precursor and uptake decarboxylase system; they have catecholamine-containing granules and can be identified immunohistochemically by staining with neuron-specific enolase, chromogranin A, and synaptophysin. Type II, known as supporting or sustentacular cells, stain positive for S-1000 and glial fibrillary acidic protein.2
Nonchromaffin paragangliomas are notoriously hypervascular with well-defined edges and a pseudocapsule. Areas of necrosis or hemorrhage are not typically seen except in cases of malignancy.2 Histologically, they appear benign with predominantly type I cells, but also type II cells and capillaries. They have nests of round, polygonal, or spindle-shaped epithelioid cells surrounded by an elaborate vasculature.28 Nuclear atypia is variable and does not correlate with clinical behavior. Figure 23-1 shows the typical histologic appearance of a paraganglioma. Head and neck paragangliomas are almost always negative for chromaffin reaction, which is not sensitive enough to identify the small amounts of catecholamines they contain. Alternatively, immunohistochemical staining for neuron-specific enolase, chromogranin A, synaptophysin, and serotonin can be used to identify type I cells and S-100 and glial fibrillary acidic protein staining can identify type II cells.2,28
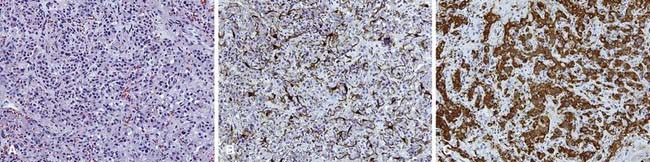
Malignancy is typically diagnosed clinically, as there are no established pathologic criteria for diagnosis. Attempts have been made to use mitoses, nuclear polymorphism, or capsular invasion to reliably predict metastatic potential.29 Others have shown that most carotid-body tumors exhibit capsular invasion,30 whereas some metastatic carotid-body tumors do not contain mitoses.31
Clinical Presentation
Carotid-body tumors generally present as a slow-growing, palpable neck mass at the bifurcation of the common carotid artery that has been present for several years.32 Other symptoms or signs may include neck discomfort, true-vocal-cord immobility that may result in hoarseness (from cranial nerve X involvement), tongue weakness and atrophy (from hypoglossal nerve XII involvement), or Horner syndrome (from involvement of the sympathetic chain).14 A very large mass may displace the soft tissue of the airway, resulting in airway symptoms like stridor extension into the parapharyngeal space that may rarely lead to dysphagia. Carotid sinus syndrome has also been reported, as in bilateral neck involvement.
Glomus-vagale tumors are less common than carotid-body tumors and appear more cephalad in the neck. They present as an upper lateral neck mass, often visible only intraorally, displacing the oropharynx anteromedially. Involvement of cranial nerves X and XII or Horner syndrome, as described in carotid-body tumors and accessory nerve weakness, are present in approximately 50% of cases.7
Glomus-tympanicum tumors usually arise in the tympanic cavity or floor of the middle ear, and are typically small and well circumscribed. They may appear as a blue or reddish lesion behind the tympanic membrane. The most common symptoms are progressive conductive hearing loss and pulsatile tinnitus.33 Other symptoms include aural pressure or fullness, vertigo, and headache.
Routes of Spread
Most paragangliomas are benign, but may be locally destructive, invading into bone and displacing or invading adjacent soft tissue. Carotid-body paragangliomas may affect adjacent cranial nerves X and XII and the sympathetic chain. Temporal-bone paragangliomas may cause destruction of the temporal bone and posterior cranial fossa. They may extend medially to the internal auditory canal and involve cranial nerves VII and VIII. Laterally, they may progress to or through the tympanic membrane and appear as a middle-ear mass or a mass in the external auditory canal. It has been reported that 5% of temporal bone and 10% to 15% of carotid-body and vagale paragangliomas are malignant, determined clinically rather than histologically.34,35 Malignant paragangliomas can spread to lymph nodes or distant sites. Caution must be taken to avoid confusing multifocality with metastases or extensive growth, cranial nerve involvement, or recurrence as a sign of malignancy.
Diagnostic Studies
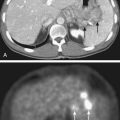
Paragangliomas are often diagnosed presumptively by their typical radiographic appearance rather than by fine-needle aspiration (FNA) or open biopsy, which are associated with the risk of bleeding. Radiographic evaluation also details the extent of the lesion and its relationship to adjacent critical structures. Because paragangliomas are hypervascular, they typically appear as strongly enhanced and well circumscribed on magnetic resonance imaging (MRI) or contrast-enhanced computed tomography (CT). If there is skull-base bone involvement, a high-resolution temporal bone CT will supplement the MRI in delineating tumor extent at the eroded jugular fossa (Fig. 23-2A and Fig. 23-2B) and through the numerous vascular channels and bony foramina.
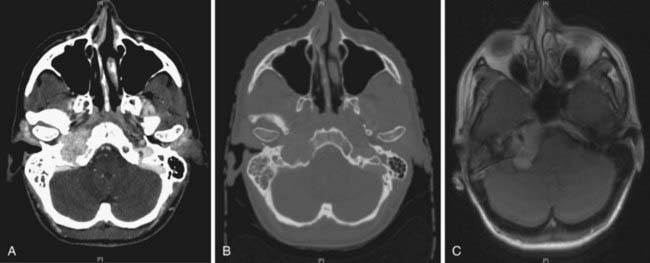
MRI provides important information about tumors and adjacent soft tissues and vascular channels. Flow voids within tumors7 produce the characteristic “salt and pepper” appearance commonly seen on a T2-weighted MRI sequence, representing areas of hemorrhage, slow-flowing blood, and tumor cells.36 MRI is superior to CT in determining skull-base involvement, intracranial extension, dural sinuses, and encasement of the internal carotid artery and internal jugular vein,7,37,38 although CT better determines middle- and inner-ear involvement and bony erosion of the jugular fossa or temporal bone.7 Figure 23-2C shows an MRI of a glomus jugulare.
Ultrasound has been used in the diagnosis of carotid-body or vagale tumors, but is of limited value for temporal-bone tumors and provides much less information than MRI. The classic appearance is that of a hypoechoic and heterogenous hypervascular lesion. For carotid-body tumors, splaying of the internal and external carotid artery may be seen.39 A Doppler-flow study showing upward movement of intratumoral blood flow at the carotid bifurcation is diagnostic of a carotid-body tumor, whereas downward flow of a neck mass indicates a glomus vagale.40
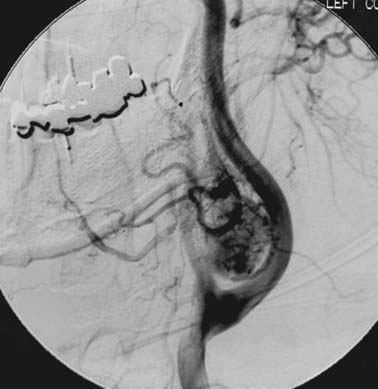
Angiography will confirm the hypervascularity of these tumors and also is an option to detect multifocality. Figure 23-3 shows a typical angiogram with intense tumoral vascularity and splaying of the internal and external carotid arteries.
A family history of paragangliomas requires a more extensive radiographic workup because of the higher likelihood of multifocal disease. Because paragangliomas share similar histologic features with other neuroendocrine tumors, including somatostatin receptors, octreotide scintigraphy may be another way to detect multifocality (or metastases).41,42 Radiolabeled metaiodobenzylguanidine, an analog of norepinephrine, is taken up and stored in the intracellular vesicles of neuroendocrine tumors and can be useful for diagnosing tumors of neuroendocrine origin, such as neuroblastoma,43 pheochromocytoma,44 and other paragangliomas.45
Staging
There is no universally accepted staging system for carotid-body and glomus-vagale tumors, but several have been published for temporal-bone paragangliomas. Some authors have used the staging system proposed by McCabe and Fletcher.46 The Glasscock-Jackson system is also available.47 The classification by Fisch48 is often used for surgical outcomes.
Standard Therapeutic Approaches
Radiation and surgery can be used as definitive treatment for paragangliomas. When the entire tumor cannot be removed, postoperative radiation is usually recommended. The decision of which modality to use is based on a number of factors such as patient age and overall health, and on the potential short-term and long-term risks of treatment complications. In general, surgery is preferred when complete resection is anticipated with a minimal surgical risk and in younger patients. This is especially so for early-stage, temporal-bone tumors, including middle-ear tumors and small-volume neck tumors that have a low attendant risk of permanent postoperative complications. Larger and locally invasive tumors may be treated with postoperative irradiation, or may be treated with primary radiotherapy because of potential cranial nerve or vascular injury associated with resection. When treated with irradiation, paragangliomas usually remain stable or may regress slowly, resulting in a persistent radiographic or palpable mass.49 Hence local control is defined as the lack of tumor progression. Symptoms caused by the tumor often improve with radiation.
Surgery
The role of preoperative embolization is debated.17 Advocates argue that surgical blood loss is decreased and operative time shortened. Critics cite the potential morbidity of angiography and the possible distortion of tissue planes during dissection. In addition, there is a small risk of embolization of an unintended vessel, and numerous reviews fail to show a perioperative benefit in reducing blood loss.50
It is advisable to obtain proximal and distal (caudal and cephalad) control of the internal carotid artery, external carotid artery, and common carotid artery prior to extensive dissection of the tumor itself. Resection of the external carotid artery at times may be required to facilitate this.51 Similarly, caudal identification of the cranial nerves X, XI, and XII, as well as the sympathetic chain facilitates their safe dissection from the tumor during subsequent periadventitial (sometimes referred in the literature as subadventitial) dissection of the paraganglioma. Nerves macroscopically involved by tumor are best resected to achieve the 90% to 95% complete resection often reported.
Chemotherapy
The role of chemotherapy for paragangliomas remains undefined, and is usually reserved for rare metastatic disease and unresectable postirradiation recurrences. Solitary case reports have demonstrated activity for several agents, including carboplatin, cisplatin, cyclophosphamide, gemcitabine, and etoposide.52–55 Some series have reported combination therapy with cyclophosphamide, vincristine, doxorubicin, and dacarbazine (DTIC),56 as well as with cyclophosphamide, cisplatin, doxorubicin, and DTIC with mixed results.57
Somatostatin analogs58 and radiopharmaceuticals based on somatostatin analogs such as octreotide and lanreotide have been evaluated because of the expression of somatostatin receptors on paragangliomas. Several radiopharmaceuticals have been applied, including 111In-pentetreotide/111In-DOTA octreotide, 90Y-DOTA-octreotide and 177Lu-DOTA-octreotate, and 111In and 90Y DOTAlanreotide.59–61
A molecularly targeted approach using imatinib mesylate, a selective inhibitor of the ABL, platelet-derived growth factor receptor (PDGFR), and stem-cell-ligand receptor (c-kit) tyrosine kinases, in 15 adult patients with disseminated endocrine tumors did not prove to be effective.62 Based on preclinical data, other strategies (e.g., targeting RAF) are being explored, but clinical evaluation remains to be performed.63
Simulation and Treatment Planning
Intensity-modulated radiation therapy (IMRT) can provide a highly conformal dose distribution that decreases the dose to surrounding normal tissues (Fig. 23-4). These techniques can be especially important for glomus-jugulare and tympanicum tumors to spare the adjacent cochlea. Although the dose normally required for these tumors is moderate, minimizing the integral dose is an important goal to reduce these long-term risks. Acute toxicities can also be reduced by limiting the dose to oral cavity structures and salivary glands. Diagnostic information that can help in defining the gross tumor volume (GTV)64 should be reviewed, including CT, MRI, and angiograms. The GTV encompasses the visible gross tumor on diagnostic imaging scans. The clinical target volume (CTV) only requires a small margin of approximately 0.5 cm to account for microscopic extension. Regional lymph-node spread is rare, so the regional nodes are not electively included in the target volume unless malignancy is suspected. The planning target volume (PTV), taken into account for set-up uncertainty and tumor motion, typically adds another 0.5 cm. If there is evidence of lymph-node spread (malignancy), the regional nodes are included in the CTV. The PTV may be extended slightly superiorly and inferiorly if the tumor extent is indistinct on the planning scan. Sensitive surrounding critical normal structures should be contoured to avoid exceeding normal-tissue tolerances, including the globes, brainstem, salivary glands, and bilateral cochleas.
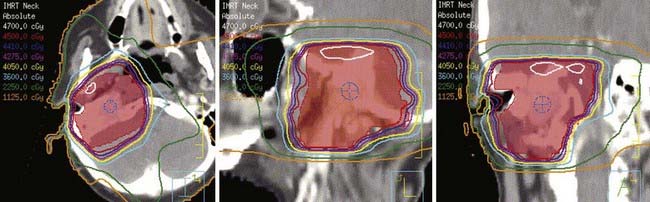
FIGURE 23-4 • Intensity-modulated radiation therapy plan showing conformality of isodose curves around the planning target volume (red).
Moderate doses of 45 to 50 Gy at 1.8 to 2 Gy per fraction adequately control benign paragangliomas without exceeding the tolerances of adjacent neurologic and optic structures such as the spinal cord, brainstem, and optic chiasm. Local control appears to be reduced with doses lower than 40 Gy.65
Stereotactic Radiosurgery
The use of stereotactic radiosurgery (SRS) to treat temporal-bone tumors is increasing, because it can deliver a large dose of radiation in few fractions and minimize normal-tissue doses.66–71 Treatment planning should again use contrast-enhanced CT, MRI, and angiography to help delineate tumor extent. A variety of doses ranging from 15 to 27 Gy in a single fraction have been published in the literature. It appears that 15 to 18 Gy in a single fraction adequately controls the disease and minimizes the risk of complications.66–71
Outcomes
Results with any treatment approach are excellent. Table 23-1 and Table 23-2 show that the local control rates of selected patients treated with surgical resection and radiation are comparable, typically greater than 85% to 95%.72 Local control in radiation series is generally defined as lack of tumor progression after treatment. Although there is less data for carotid-body tumors, the available data suggests that control rates equal those for temporal-bone tumors. Valdagni and Amichetti reported on 7 patients with 13 carotid-body tumors treated with radiation and found 100% local control.73 Verniers and colleagues reported a series including 17 carotid-body tumors, none of which recurred after radiotherapy, with a mean follow-up of 10 years.74 In a series reported by Hinerman and colleagues, 18 patients with 25 carotid-body tumors or glomus-vagale tumors were treated with radiotherapy. The 15-year local control rate was 92%.75
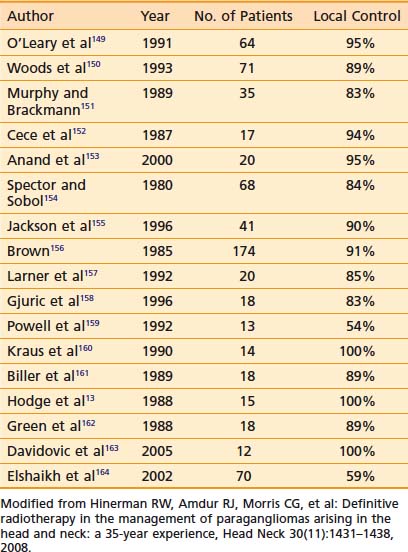
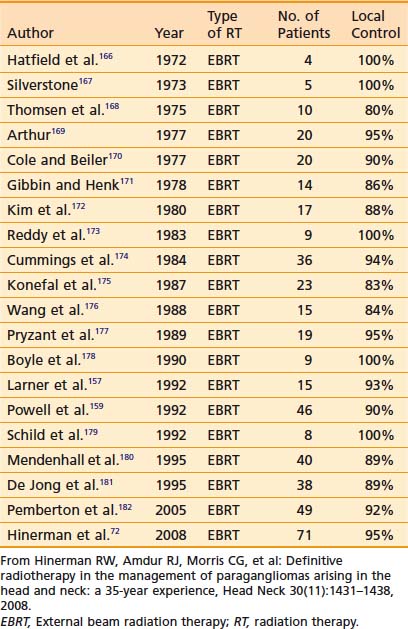
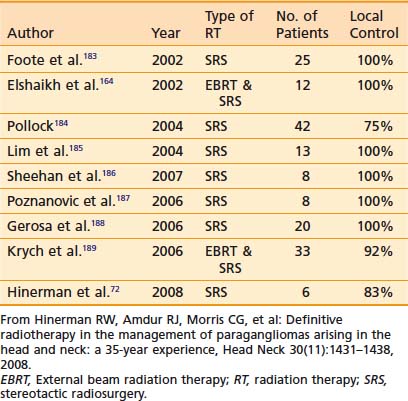
Because SRS is relatively new, less data on treatment outcomes is available and follow-up is shorter. Early results show excellent local control (Table 23-3). However, because this disease can have late failures, longer follow-up is needed to show results comparable to surgery or fractionated radiotherapy.
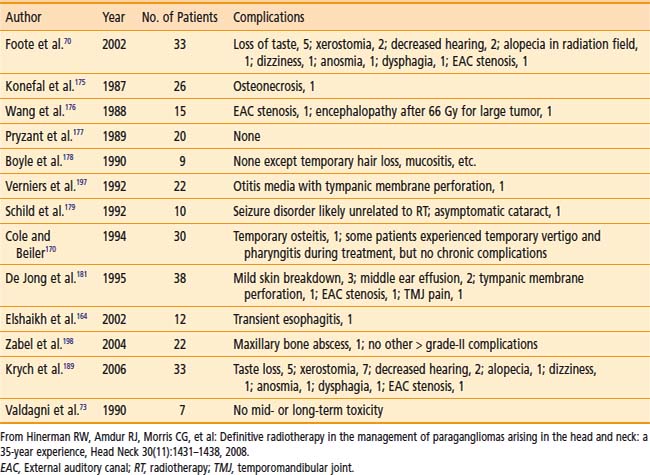
Treatment Toxicity
Table 23-4 shows surgical complications during the preceding two decades. Most surgical morbidity is related to cranial-nerve deficits, which can be either temporary or permanent. Other complications are related to wound infections or vascular damage. With appropriate selection and modern surgical and anesthesiologic methods, permanent cranial-nerve deficits are approximately 10% to 15%, and vascular injury is rare. Isolated X or XII deficits are well tolerated, and compensatory rehabilitation is available with good results when there are combined deficits.72,76
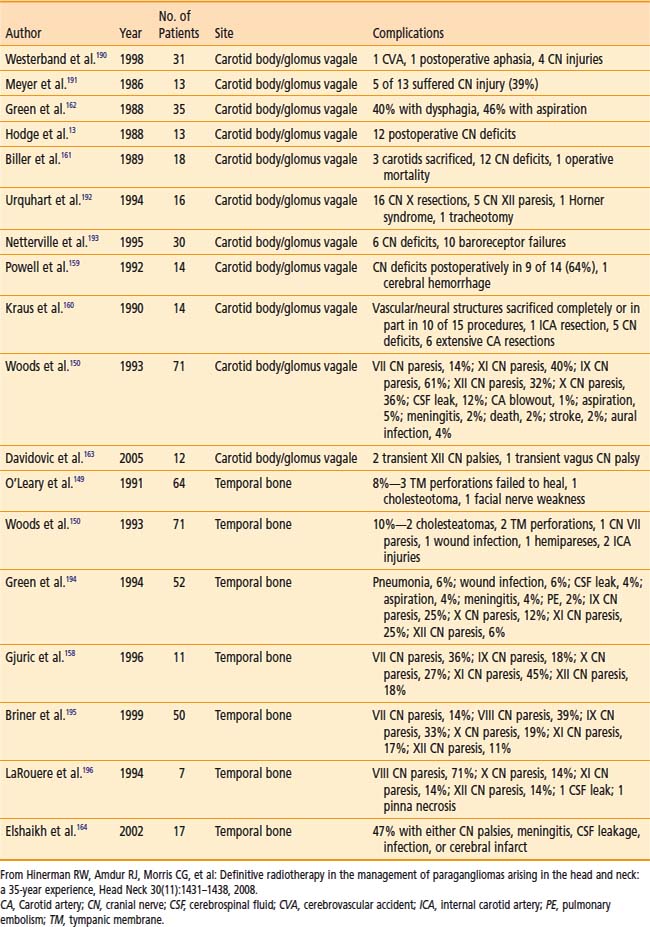
Table 23-5 shows the reported treatment-related morbidity with radiotherapy. Cranial-nerve deficits and bone necrosis are occasionally seen after radiotherapy. Late radiation-induced malignancy, though possible, has not been reported. It is hoped that complication rates will continue to improve as imaging and radiation delivery technologies improve.
Chordoma and Chondrosarcoma
In a study that extracted data from the Survival, Epidemiology, and End Results database, McMaster and colleagues found that 32% of chordomas arise in the skull, 33% in the spine, 29% in the sacrum, and only 6% elsewhere.77 Noel and colleagues examined 47 cases of chordomas in the skull or cervical spine. Of these, 23 tumors (49%) arose in the clivus, 15 (32%) in the sphenoclival region, 4 (9%) in the petroclival region, 3 (6%) in the cervical spine, and 2 (4%) in other areas.78 They observed an incidence of 0.08 patients per 100,000. The incidence of chordoma has been stable during the preceding three decades.77 The median age of diagnosis for chordomas is in the sixth to seventh decade.79 There is a slight male predominance, and the disease rarely occurs in blacks.79
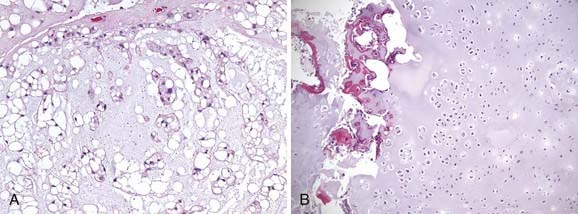
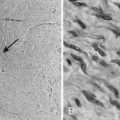
Histologically, chordomas appear as benign cells, but their clinical behavior belies that microscopic appearance. They were first described microscopically as physaliferous cells because of their large vacuoles.79 The cells are uniform in appearance with small, oval nuclei containing dense chromatin (Fig. 23-5A). There are three subtypes of chordomas: (1) classic, (2) dedifferentiated, or (3) chondroid. The classic and chondroid variants are the most commonly seen subtypes in skull-base tumors. Chondroid chordoma has been thought to carry a better prognosis than chondrosarcoma, but they are often difficult to distinguish. Data also suggest that the outcomes for the chondroid and classic variants are similar.80,81 In comparison, dedifferentiated chordomas are more aggressive, with more mitotic activity and cellular atypia, similar to a round-cell tumor or spindle-cell sarcoma. Dedifferentiation occurs in fewer than 4% of cases at presentation82 and most commonly occur in the sacrococcygeal region.79
Genetic changes linked to the development of chordomas have been mapped to chromosomes 1p, 3p, and 7q. Scheil and colleagues determined that mismatched repair genes on 1p and 3p as well as oncogenes found at 7q may be involved in the development of chordomas.83

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


