- 11.1 Introduction
- 11.2 Infections and the skin
- 11.3 T-cell-mediated skin disease
- 11.3.1 Contact dermatitis
- 11.3.2 Atopic eczema
- 11.3.3 Psoriasis
- 11.3.1 Contact dermatitis
- 11.4 Autoimmune skin disease
- 11.4.1 Bullous skin disease
- 11.4.2 Vitiligo
- 11.4.3 Alopecia areata
- 11.4.1 Bullous skin disease
- 11.5 Systemic diseases with skin involvement
- 11.5.1 Cī inhibitor deficiency
- 11.5.2 Vasculitis
- 11.5.3 Cryoglobulinaemia
- 11.5.4 Lupus erythematosus
- 11.5.5 Systemic sclerosis
- 11.5.1 Cī inhibitor deficiency
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
11.1 Introduction
The skin consists of an impervious horny layer of stratified squamous epithelial cells called keratinocytes (epidermis) overlying vascular connective tissue (dermis). The epidermis has an important mechanical function as a barrier from the outside world, but also plays a more active role in the skin’s response to injury. The keratinocyte can synthesize a large number of cytokines and other inflammatory mediators in response to injury or ultraviolet radiation (Fig. 11.1). These non-specific mediators increase vascular permeability, attract and immune activate cells immune and induce the expression of adhesion molecules on nearby endothelial cells to allow these cells access to the damaged tissue.
Fig. 11.1 Multiple proinflammatory factors produced by keratinocytes in response to injury. IL, Interleukin; MHC, major histocompatibility complex; TNF, tumour necrosis factor; TGF, transforming growth factor.
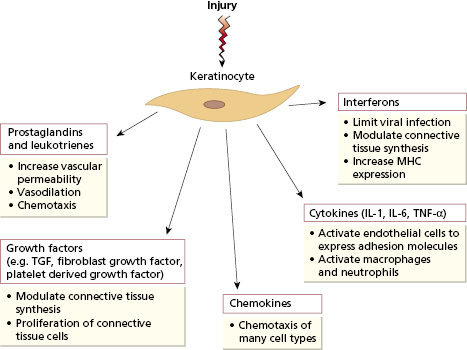
Intertwined with this innate response to injury is a system for adaptive immunity against antigens gaining access via the skin. Around 10% of the cells in the epidermis are a specialized antigen-presenting population known as Langerhans cells. These cells have a high capacity for the uptake and processing of antigen. Following any insult to the epidermis (such as invasion by microorganisms), they migrate along afferent lymphatics to the paracortex of the draining lymph nodes; during migration they differentiate into classical dendritic cells with high expression of major histocompatibility complex (MHC) class II and co-stimulatory molecules (see Chapter 1, section 1.3.4). They then activate specific naive T cells, via TCRs for those peptides derived from antigens taken up in the skin, inducing proliferation and differentiation of T cells into effector cells. Most of these leave the lymph node, enter the blood and then migrate into inflamed skin. In order to return to the particular site of inflammation, T cells express receptors (integrins) that are specific for intercellular addressin cell adhesion molecules (ICAMs) found on endothelial cells in inflamed skin (the expression of which is induced by cytokine signals from keratinocytes). The best characterized T-cell adhesion molecule for homing to skin is known as cutaneous lymphocyte-associated antigen (CLA), which binds to E-selectin on endothelial cells. Skin-homing T cells also express the chemokine receptors CCR4 and CCR10, which selectively bind a distinctive chemokine produced by cutaneous endothelial cells and keratinocytes. This leads to migration and retention of these T cells in the skin. Langerhans cells, keratinocytes, CLA-positive T lymphocytes and local lymph nodes have been regarded collectively as skin-associated lymphoid tissue (SALT). Although this tissue limits infection, it is also responsible for some types of skin disease (see section 11.3).
Unlike the rest of the body, the skin is exposed to ultraviolet radiation (UVR), which has important local and systemic immunological effects (Table 11.1). Chronic UVR exposure leads to skin cancer due to oncogenic human papilloma viruses (HPV). The risk of skin malignancy is increased in immunosuppressed patients, especially in skin exposed to high levels of UVR. This suggests that immune mechanisms normally control HPV infection. Particular strains of HPV produce E6 and E7 proteins that effect cell cycle components and can lead to reduced apoptosis and eventually uncontrolled growth, i.e. malignancy, if not checked by immune responses. UVR exposure depresses Langerhans cell function, reduces in vitro lymphocyte responses and impairs cell-mediated immunity (Table 11.1 and Fig. 11.1). Infection, autoimmunity or hypersensitivity can cause skin diseases. Skin damage may be triggered by autoantibodies to skin antigens (in the bullous diseases) or by deposition of immune complexes [in systemic lupus erythematosus (SLE)]. T cells are involved in some forms of dermatitis.
Table 11.1 Effects of ultraviolet radiation on the skin
| Induces skin cancer formation |
Effect on Langerhans cells
|
Effect on lymphocytes
|
11.2 Infections and the skin
Bacterial, viral and fungal infections can occur when the physical barrier of the skin is breached following trauma (especially burns) or widespread eczema, or if immune defences are impaired by systemic or topical immunosuppressive treatment due to exposure to corticosteroids and cytotoxic drugs (Chapter 7) or due to primary or secondary immunodeficiency (see Chapter 3). Septicaemic infections, e.g. gonococcal or meningococcal septicaemia, may seed foci of infection into the skin. Certain viruses invade the skin to produce infected lesions [chickenpox (VZV) or warts (HPV)], although a reactive, non-infective rash is a more common response to systemic viral infections (e.g. exanthemata of rubella or measles). Recurrent herpes labialis is caused by reactivation of persistent herpes simplex infection; attacks are often provoked by exposure to UV light, probably due to suppression of the skin immune system (Case 11.1). Atopic individuals have an increased tendency to herpes labialis, perhaps related to a less effective T-cell response against this virus. Skin granulomas, although rare in developed countries, are most commonly caused worldwide by invading microorganisms such as Mycobacterium tuberculosis, M. leprae or Treponema pallidum.
 Case 11.1 Recurrent cold sores
Case 11.1 Recurrent cold soresA 38-year-old woman had been troubled since the age of 8 by recurrent cold sores. Several times each year she would develop a distinctive tingling sensation around her nose or lips, followed several hours later by localized formation of small blisters which crusted, became more painful and gradually cleared over several days. The attacks were often provoked by exposure to strong sunlight. She had a history of troublesome hay fever but was otherwise well. She was able to prevent attacks by use of a high-factor sun-block and treat any breakthrough episodes with prompt use of aciclovir cream at the onset of symptoms.
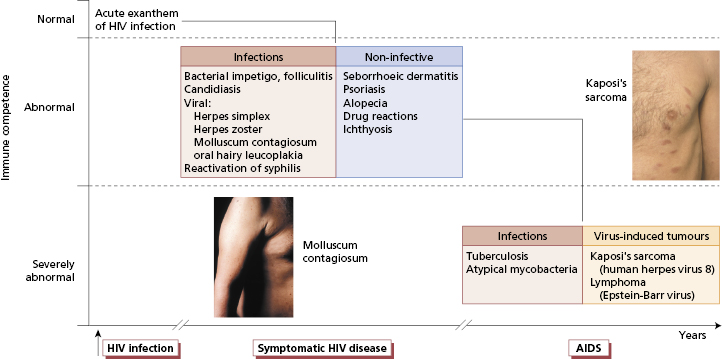
One of the best examples of the multiple skin manifestations associated with immunodeficiency is human immunodeficiency virus (HIV) (see Chapters 2 and 3). A wide spectrum of skin problems occurs in HIV-infected individuals (Fig. 11.2). Sometimes these are exacerbations of pre-existing skin disease, such as psoriasis, but more often they are new conditions.
Fig. 11.2 The spectrum of skin disease in human immunodeficiency virus (HIV)-positive individuals. The disorder on the left is molluscum contagiosum and that on the right is early Kaposi’s sarcoma.
11.3 T-cell-mediated skin disease
T cells play a central role in some of the most common skin diseases: the best understood are contact dermatitis, mediated by Th1 cells, and atopic eczema, mediated largely by Th2 cells. The chronic skin disease psoriasis also appears to be mediated largely by T cells, although the triggering antigen and the mechanism by which T cells induce the characteristic epidermal changes of psoriasis are not fully understood.
11.3.1 Contact dermatitis
Contact dermatitis is an inflammatory skin disease caused by Th1-cell-mediated (type IV) hypersensitivity to external agents that come into contact with the skin. It is an important cause of occupational skin disease. The range of potential sensitizing antigens is enormous but, fortunately, a relatively small number of substances account for most cases (Table 11.2). These agents are usually of relatively low molecular weight (<1 kDa) and are not immunogenic in their own right: as small molecules they easily penetrate the epidermis and bind covalently to skin or tissue proteins to act as haptens combined with the host cells as carriers (see Chapter 1).
Table 11.2 Some agents responsible for allergic contact dermatitis
| Agent | Examples of exposure | |
|---|---|---|
| Metals | Nickel | Clasps, necklaces, watch-straps |
| Chromate | Cement (building site workers) | |
| Cobalt* | ||
| Medications | ‘Para’-group chemicals | Benzocaine-type anaesthetics, sulphonamide antibiotics, PABA-containing substances (e.g. sunscreens) and oral hypoglycaemic agents(sulphonylureas) |
| Phenothiazines | Phenothiazine-based antihistamines | |
| Neomycin | Topical antibiotics | |
| Plastics | Epoxyresins, acrylates | Construction industry, glues |
| Rubber | Accelerators | Tyre industry, rubber gloves, shoes, clothing, household ‘grips’, etc. |
| Plants | Poison ivy (USA only) | |
| Primula | ||
| Chrysanthemum | ||
| Geranium | ||
| Cosmetics | Perfumes | |
| Preservatives | ||
| Lanolin |
*Source of cobalt sensitivity is usually obscure but it may exist as a co-sensitivity with nickel (metal) or chromate (cement).
PABA, Para-amino benzoic acid.
The diagnosis of contact dermatitis depends on a careful medical history, the distribution of the lesions and patch testing. In the patch test, the suspected contact sensitizer is applied to normal skin (usually on the upper back) and covered for 48 h. The reaction is read after 2 and 4 days. In a positive response, there is inflammation and induration at the test site. Although there are pitfalls in interpretation, patch-testing is indispensable in the investigation of allergic contact dermatitis (Case 11.2).
 Case 11.2 Nickel dermatitis
Case 11.2 Nickel dermatitisA 47-year-old woman presented with a 3-week history of an acute rash that started beneath her watch. Two weeks later, a further patch appeared at the umbilicus. She had previously noted that she could not wear cheap earrings without triggering a rash on her ear lobes. There was no past medical history of note and no personal or family history of atopy. On examination, two patches of dermatitis were seen over the presenting areas. The appearances were suggestive of nickel-induced contact dermatitis corresponding to nickel in the watch and on a jeans stud. She was patch-tested to a battery of commonly implicated agents (Table 11.2): strongly positive results were induced by nickel sulphate and cobalt chloride only. The final diagnosis was nickel dermatitis, which cleared spontaneously following avoidance of nickel-containing articles.
Contact dermatitis is a prototype of T-cell-mediated hypersensitivity (see Box 11.1). Two phases of pathogenesis are recognized: an induction phase, from the time of initial antigen contact to sensitization of T lymphocytes, and an elicitation phase, from antigen re-exposure to the appearance of dermatitis. In the induction phase, Langerhans cells bind the hapten–carrier protein complex and present it, in association with MHC class II antigens, to T lymphocytes (Fig. 11.3). Induction of cellular immunity to a contact skin sensitizer can occur within 7–10 days of first contact, but it usually happens after many months or years of exposure to small amounts of antigen (Case 11.2). Individual sensitivity varies according to the nature of the chemical, its concentration and the genetic susceptibility of the person exposed. Recently the contribution of innate immunity to inflammation during the induction phase, following recognition of the sensitiser by TLRs on dendritic cells has been reported in mice. This has to be confirmed in humans, as does the role of natural killer (NK) cells that are found in significant numbers in biopsies from patients with contact dermatitis.
- Association between psoriasis and HLA-C6 and -DR7
- Relationship with streptococcal infection, which is known to cause other immunologically mediated hypersensitivity disorders
- Worsening of psoriasis in human immunodeficiency virus infection
- Response of psoriasis to therapeutic interventions directed against T cells (such as UV radiation, monoclonal antibodies and ciclosporin/tacrolimus, although the latter drugs also have direct effects on keratinocytes)
- Excellent responses to anti-TNF therapies
Fig. 11.3 The pathogenesis of allergic contact dermatitis. Langerhans cells bind and present the hapten–carrier protein complex to T lymphocytes (T). Subsequent re-exposure to the hapten triggers a T-cell-mediated (type IV) hypersensitivity reaction in the skin. CLA, Cutaneous lymphocyte-associated antigen.
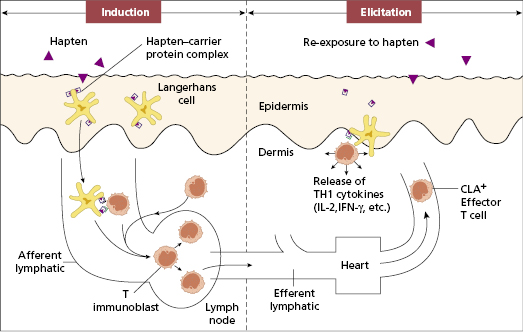
Re-exposure to the relevant antigen triggers the elicitation phase that produces characteristic features of dermatitis. In this phase, effector T lymphocytes, previously carried via the circulation to the skin, meet the hapten–carrier complex. CD8 T cells have been identified as the crucial effector population. Th1 CD4+ T cells provide activation signals through the release of cytokines such as interferon-γ and tumour necrosis factor; CD8+ and Th17 cells are activated to induce skin inflammation (Fig. 11.3), recruitment of more T cells and monocytes, keratinocyte proliferation, hyperplasia of the epidermis and consequent thickening.
The management of contact dermatitis involves two approaches: prevention and treatment. Identification and elimination of the responsible antigen is the most important goal. Unfortunately, many common antigens are ubiquitous and antigen avoidance may be difficult to achieve. Preventative measures in industrial dermatitis depend on the use of protective gloves and clothing, improved ventilation or the substitution of non-antigenic chemicals. Some medicines used to treat skin disease are among the commonest culprits of contact dermatitis (see Table 11.2); they include topical antibiotics and antihistamines. As in atopic eczema, topical corticosteroids are useful therapeutic agents, together with antibacterial measures where indicated.
11.3.2 Atopic eczema
Atopic eczema is a common disorder, occurring predominantly in childhood, which appears to be caused by barrier dysfunction with subsequent altered exposure to microbes and allergens that results in a Th2 hypersensitivity reaction in the skin. Although it is usually a mild disorder (as in Case 11.3), patients with severe atopic eczema (especially children) are really disabled and on occasions there can be life-threatening complications. The disorder and its pathogenesis are discussed in more detail in Chapter 4.
 Case 11.3 Atopic eczema
Case 11.3 Atopic eczemaA 5-year-old boy had developed an itchy rash on his trunk and feet at the age of 18 months. This waxed and waned over the next 3 years and gradually came to involve predominantly his flanks, popliteal and antecubital fossae. He had mild asthma requiring occasional bronchodilators only. His mild atopic eczema was treated with bland emollient creams and occasionally 1% hydrocortisone. His prognosis was good and his skin problems resolved over the next few years.
11.3.3 Psoriasis
Psoriasis is a common skin disease, affecting about 2–4.7% of the world’s population. It is less common in sunny climates and in those with pigmented skins. Important risk factors include smoking and a high body mass index. It can present at any age but most commonly appears between the ages of 15 and 30 years or 50 and 60 years.
Although very varied, the usual clinical form consists of chronic, raised, red, scaly, round or oval plaques, with sharply marginated edges, mainly occurring on the knees, elbows and scalp. Chronic plaques may remain static for years, resolve spontaneously or progress to the rest of the skin (Figure 11.4), so-called erythrodermic psoriasis. The diagnosis (as for much of dermatology) is a clinical one: there are no helpful tests. The quality of life of a psoriatic patient is markedly reduced, not only due to the disfigurement but also to co-morbidities including cardiovascular disease, depression and diabetes. In addition, 2–19% of patients develop a seronegative arthropathy (see Chapter 8).
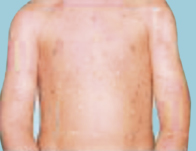
Numerous Inflammatory triggers for psoriasis have been proposed, including trauma, bacterial infections, cellular stress and various medications. However, these are not consistently observed across patients. Guttate (drop-shaped) psoriasis may begin with several small lesions 1–2 weeks after a Group A streptococcal throat infection, but these usually resolve spontaneously after a few months (Fig 11.4).
Fig. 11.4 Guttate psoriasis. Miall L, Rudolf M & Smith D (2012) Paediatrics at a Glance, 3rd Ed. Reproduced with permission from Wiley.
The pathology involves markedly increased proliferation and shedding of keratinocytes is disordered, as part of the chronic inflammation. An unknown trigger results in DNA being released from keratinocytes, forming a complex with the secreted keratinocyte cathelicidin antimicrobial protein LL-37. This acts on TLR9 on plasmacytoid dendritic cells to release type I interferons (α and β), which in turn activate myeloid dendritic cells; these cells then release IL-20 to stimulate keratinocyte proliferation. Some myeloid dendritic cells migrate to local lymph nodes, where they release IL-23 and activate naïve T cells to differentiate into Th1 and Th17 cells that are recruited back to the skin and cause inflammation via interferon-γ, IL-17 and IL-22. This results in further keratinocyte proliferation and the result is a vicious circle of keratinocytes activating dendritic cells, dendritic cells activating T cells, and T cells activating keratinocytes.
Genetic studies showed a familial trait and genome wide association studies have confirmed at least 10 psoriasis susceptibility genes that are involved in immunity, including HLA-C6 and -DR7 (Box 11.1). Polymorphisms in the IL-12/IL-13 receptor, the p40 subunit of IL-12 and IL-23, and the p19 subunit of IL-23 are strongly associated with psoriasis, supporting their critical role in the disease process and providing targets for medical therapy.
Therapy of psoriasis is aimed at breaking this cycle. Many older treatments aimed to limit keratinocyte proliferation, often using coal-tar-based drugs. Recent understanding of the role of inflammation and T cells has resulted in the use of immunosuppressant and anti-inflammatory agents. Immunosuppressant therapy may be delivered topically, for example corticosteroids or T-cell-suppressant drugs such as tacrolimus, or physically in the form of UVR. Severe disease has been transformed by the use of targeted biological therapies (Fig. 11.5), providing confirmation of the role of both T cells and TNF-α in psoriasis. This has largely replaced high doses of methotrexate or ciclosporin, the serious side effects (pulmonary fibrosis and renal disease respectively) of which are considerable. The currently approved biological therapies include the TNF-α inhibitors and ustekinumab. The TNF inhibitors, Infliximab, etanercept and adalimumab (Chapter 7
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


