Conserved catalytic domain
Activity
Localization
Principal targets under stress
Regulation by CR/ROS
Sirtuin function
References
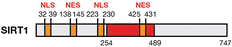
Deacetylase
Nucleus/cytoplasm
H4K9Ac, H4K16Ac, SUV39h1, XPA, XPC, NBS1, hMOF, TIP60, WRN, Ku70, Rb, FOXO, HIF-1α, p53
Up-regulated in WAT and muscle, down-regulated in liver and pancreas/up-regulated
Heterochromatin structure maintenance, ribosome biosynthesis, DNA repair, cell cycle control, apoptosis
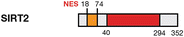
Deacetylase
Cytoplasm/nucleus
H4K16Ac, H3K56Ac, PR-SET7, CDH1, CDC20, CDK9, FOXO, p53
Up-regulated/up-regulated
Cell cycle control, apoptosis
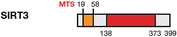
Deacetylase
Mitochondrial matrix/nucleus
AceCS2, IDH2, MnSOD, p53, Ku70
Up-regulated/Up-regulated
Metabolism homeostasis, ROS levels control, apoptosis

ADP-ribosyltransferase
Mitochondrial matrix
GDH, MCD
Down-regulated/ND
Metabolism homeostasis
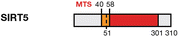
Deacetylase/deacylase
Mitochondrial matrix
CPS1
Up-regulated/ND
Metabolism homoeostasis, ROS detoxification

ADP-ribosyltransferase/deacetylase/deacylase
Nucleus
H3K9Ac, H3K56Ac, PARP1, CtiP, SNF2H, NF-kB, p53, p73
Up-regulated/ND
Telomere maintenance, DNA repair, metabolism homeostasis ribosome biosynthesis, apoptosis
Deacetylase
Nucleolus
H3K18Ac, RNA Pol, p53
Up-regulated/ND
rDNA expression, ribosome biogenesis, apoptosis
Sirtuins seem to be focused on three related forms of stress, all closely related to carcinogenesis (Yang et al. 2003): metabolic (excluding oxidative), oxidative and genotoxic (Bosch-Presegué and Vaquero 2013) stress. In metabolic stress, Sirtuins promote metabolic homeostasis at the cellular and systemic levels and are intimately linked to the endocrine system—particularly, to the insulin/insulin-like growth factor associated pathways. An interesting example of this type of stress is calorie restriction (CR), a 30–50 % reduction in food intake that has been shown to extend lifespan in many organisms and to improve human pathologies such as cancer or diabetes (Qiu et al. 2010). Some studies report that Sirtuins partially mediate the beneficial effects of CR on aging and age-related diseases in mammals.
Oxidative stress refers to the conditions generated by reactive oxygen species (ROS) produced by the mitochondrial respiratory chain, peroxisomes, lipooxygenases, radiation, chemotherapeutics or environmental toxins. Interestingly, CR indisputably reduces production of ROS (Vaquero and Reinberg 2009). Paradoxically, although CR and ROS are opposing phenomena, they both activate Sirtuins.
Finally, genotoxic stress encompasses the genomic damage caused by a harmful endogenous or exogenous agent, including metabolic products, ROS, radiation (e.g. UV or ionizing), chemotherapeutics or environmental toxins. These agents damage DNA in different ways, including single-strand breaks (SSBs) and double-strand breaks (DSBs), and drastically alter chromatin organization and structure as well as the cell cycle (Yang et al. 2003). In genotoxic stress response, Sirtuins regulate chromatin structure maintenance, DNA repair, gene expression, cell-cycle progression and apoptosis.
5.2 Activation of Sirtuins by Stress
Given their critical role in stress response, Sirtuins are activated and regulated by myriad signaling pathways. However, current knowledge on these mechanisms is chiefly restricted to SIRT1 and SIRT6. The mechanisms identified to date include gene expression, RNA stability and post-translational modifications (PTM).
5.2.1 Modulation of Sirtuin Gene Expression and RNA Stability
Expression of all seven eukaryotic Sirtuins is tightly regulated by different combinations of transcription factors. For instance, CR induces expression of SIRT1-3 and SIRT6, inhibits expression of SIRT4, and does not affect expression of SIRT5 (Qiu et al. 2010). CR activates the expression of SIRT1 in white adipose tissue (WAT) and muscle accordingly with the higher levels of NAD+ in the cytoplasm and nucleus. In contrast, it represses the expression of SIRT1 in the liver and pancreas, where, compared to the aforementioned tissue types, the NAD+/NADH ratio is lower in the nucleus and cytoplasm, and higher in the mitochondria.
Expression of SIRT1, the best-studied Sirtuin, is regulated by various transcription factors involved in cell growth, differentiation and migration, in stress resistance and in metabolism. Interestingly, most of these different forms of SIRT1 regulation exhibit a feedback loop between a given factor and SIRT1. These factors include the tumor suppressor p53, which is involved in cell-cycle checkpoint regulation, stress response and apoptosis. Two functional p53 binding sites have been identified in the SirT1 promoter. In response to cellular access to nutrients, p53 regulates SIRT1 expression in different directions. In nutrient-deprived mammalian cells, FOXO3 transcription factor forms a complex with p53, which is recruited to the Sirt1 promoter sites, stimulating SIRT1 expression (Nemoto et al. 2004). Another interesting factor is the redox sensor Carboxy-terminal of E1A-binding protein (CtBP), which can directly sense changes in the NAD+/NADH ratio and might be among the earliest acting sensors of the response. In response to metabolic stress, Sirt1 gene expression is regulated by CtBP and the tumor suppressor Hypermethylated in cancer-1 (HIC1). Cellular redox changes sensed by CtBP alter its affinity for HIC1, thereby diminishing CtBP recruitment to the Sirt1 promoter and derepressing Sirt1 expression (Zhang et al. 2007). In turn, SIRT1 interacts with and deacetylates HIC1, enabling its sumoylation and increasing its transcriptional repression activity (Stankovic-Valentin et al. 2007). In contrast, under normal nutrient conditions, p53 mediates Sirt1 gene repression in cooperation with HIC1 (Chen et al. 2005).
Another important regulator of SIRT1 gene expression is E2F1, an oxidative stress and DNA damage-response transcription factor that controls G1/S phase progression. E2F1 binds directly to the SIRT1 promoter and, in cells treated with the topoisomerase II inhibitor etoposide, upregulates expression of SIRT1. Phosphorylation of E2F1 by the stress-response kinase Ataxia telangiectasia mutated (ATM) seems to be necessary for E2F1-mediated regulation of Sirt1 gene expression. In turn, SIRT1 interacts with and deacetylates E2F1, inhibiting its pro-apoptotic activity and inducing cell proliferation (Wang et al. 2006). Another feedback of SIRT1 has been reported with the oncogene c-Myc, which upregulates Sirt1 expression. In turn, SIRT1 deacetylates c-Myc, leading to its degradation and inhibiting cellular proliferation (Yuan et al. 2009).
Interestingly, E2F1 and p53 also regulate SIRT1 at the translational level. The microRNAs miR-34a and miR449a, which are targets of p53 and E2F1, respectively, both inhibit SIRT1 expression (Yamakuchi et al. 2008). Moreover, SIRT1 cellular levels are also regulated by the RNA binding protein HuR, which associates with SIRT1 mRNA, leading to increased Sirt1 mRNA stability and consequently, to increased levels of SIRT1 protein (Abdelmohsen et al. 2007).
SIRT6 is a histone deacetylase and mono[ADP-ribosyl]transferase involved in different mechanisms of genome protection during stress. Under nutritional stress, SIRT1 forms a complex with FOXO3a and Nuclear respiratory factor (NRF)1 at the SIRT6 promoter to positively regulate SIRT6 expression (Kim et al. 2010).
5.2.2 Post-translational Modification
As previously stated, the other major mechanism for regulating Sirtuin function is PTM, which can occur at different levels. For instance, in response to oxidative stress, JNK kinase phosphorylates and stimulates SIRT1. In contrast, under stress, mTOR phosphorylates and inhibits SIRT1, suggesting a complex regulatory mechanism that could temporally regulate SIRT1 activity (Back et al. 2011; Nasrin et al. 2009). Moreover, under stress different kinases phosphorylate SIRT1 at different residues, modifying its p53 deacetylation capacity. For example, phosphorylation of SIRT1 by DYRK1A or DYRK3 stimulates it to deacetylate p53 (Guo et al. 2010), and by AMPK, inhibits its capacity to deacetylate p53 (Lee et al. 2012). SIRT1 undergoes other PTMs besides phosphorylation. For instance, in response to DNA damage, it is methylated by Set7/9, which inhibits its ability to deacetylate p53 (Liu et al. 2011). Alternatively, under genotoxic stress, sumoylation of SIRT1 by SUMO1 increases its activity, whereas desumoylation of SIRT1 by SENP1 reduces its catalytic activity (Yang et al. 2007).
SIRT2, SIRT6 and SIRT7 are also modified by phosphorylation, mainly during mitosis. The roles of these modifications (e.g. SIRT2 P-S368 or SIRT6 P-S303) are poorly understood, although they appear to relate to mitotic progression. In the case of SIRT6, phosphorylation of residue S338 has been suggested to participate in protein interaction (Dephoure et al. 2008; Miteva and Cristea 2014; North and Verdin 2007). Finally, another interesting modification in SIRT6 is its auto ADP-ribosylation, although its role also is unknown (Liszt et al. 2005).
5.3 Sirtuins in Stress Response
As previously mentioned, Sirtuins coordinate stress response at many levels, which can be divided into four groups: genome stability, regulation of gene expression, metabolism and apoptosis and senescence.
5.3.1 Genome Stability
Maintenance of genome integrity is essential for preventing tumorigenesis and ensuring cell viability. Sirtuins regulate genome stability via three principal processes: by regulating chromatin, through modulation of its structure and dynamics; by participating in DNA repair pathways; and finally, by regulating cell–cycle progression, which apparently includes direct involvement in checkpoint regulation (Fig. 5.1).
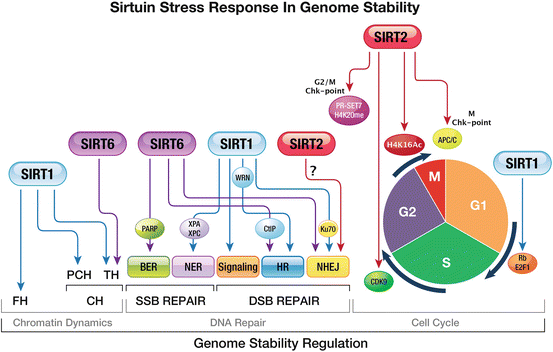
Fig. 5.1
Response of Sirtuins to stress: ensuring genome stability. Sirtuins are keystones of cellular stress response, regulating genome stability via three main cellular processes: chromatin dynamics DNA repair and cell–cycle progression. Chromatin dynamics: SIRT1 and SIRT6 are the only Sirtuins that regulate heterochromatin formation. Under stress, SIRT1 promotes formation of FH and CH at pericentromeric and telomeric regions. However, SIRT6 only promotes formation of TH. DNA repair (SSB and DSB): Sirtuins have been imputed in DNA-repair signaling and response. SIRT6 activates BER by mono(ADP-ribosyl)ating PARP1 and SIRT1 activates NER via up-regulation of the repair enzymes XPA and XPC. SIRT1, SIRT6 and SIRT2 are important for DSB repair. In HR SIRT6 promotes DNA-end resection by regulating CtIP and SIRT1 activates the helicase WRN. SIRT1 induces NHEJ by deacetylating Ku70, thereby inducing Ku70-dependent DNA repair. Cell-cycle progression: the main Sirtuin involved in cell-cycle regulation is SIRT2. During the G2/M transition, SIRT2 deacetylates H4K16Ac and the HMT PRSET7, enabling deposition of H4K20me1 by the latter. In metaphase, SIRT2 activates APC/C, enabling mitotic exit. SIRT1 promotes cell proliferation by regulating Rb/E2F1. Abbreviations: BER base excision repair, Chk-point checkpoint, CH constitutive heterochromatin, FH facultative heterochromatin, HR homologous recombination, NER nucleotide excision repair, NHEJ non-homologous end joining, PCH pericentromeric heterochromatin, TH telomeric heterochromatin
5.3.1.1 Regulation of Chromatin Structure and Dynamics
Regulation of chromatin structure is among the most important of the stress responses mediated by Sirtuins. However, only SIRT1 and SIRT6 are known to have this function, which they perform by deacetylating conserved histone marks and by modulating other chromatin factors, including transcription factors and chromatin enzymes.
Facultative Heterochromatin (FH)
In response to stress, SIRT1 acts as an expression silencer: it promotes FH formation, which results in silencing of specific genes linked to specific pathways in the context of stress response (see below). In fact, SIRT1 promotes epigenetic silencing of the targets regulated by major transcription factors, by coordinating several events together with other enzymes (Vaquero et al. 2007). Chromatin immunoprecipitation experiments have shown that when SIRT1 is recruited to a euchromatin region, there is a reduction in the euchromatin marks H3K79me2, H4K16Ac and H3K9Ac; recruitment and deacetylation of histone H1; and an increase and spreading of heterochromatin marks such as H3K9me3 and H4K20me1 (Vaquero et al. 2004). In this sense, SIRT1 promotes the spreading of the repressive mark H3K9me3, through its functional relationship with the histone methyltransferase (HMT) SUV39h1, the main H3K9me3 activity in mammals. SIRT1 interacts with, recruits and deacetylates SUV39h1 at its catalytic domain, thereby increasing SUV39h1 activity (Vaquero et al. 2007).
Another example of SIRT1 in FH occurs during development. SIRT1 levels are high in non-differentiated cells and decrease with increasing differentiation. This is interesting in the development of tissue types that are particularly sensitive to energy or redox fluctuations, such as brain tissue. SIRT1 is part of Polycomb Repressive Complex 4 (PRC4), which contains the H3K27me3 HMT Ezh2 (Kuzmichev et al. 2002). PRC4 activity results in methylation of H1K26 through previous deacetylation of H1K26 by SIRT1. This in turn would create a binding site for heterochromatin protein (HP) 1, thereby promoting heterochromatinization (Vaquero 2009). Consistently with this scenario, all subunits of the PRC complexes, including SIRT1, are overexpressed in breast, colon and prostate cancers (Chang and Hung 2012). SIRT1 also interacts with the histone H3K4 demethylase LSD1/KDM1A complex (Mulligan et al. 2011). This complex helps repress genes governed by the Notch signaling pathway, via H4K16Ac deacetylation and H3K4me1/2 demethylation. Said pathway is critical in mammalian development and is involved in tumorigenesis (Mulligan et al. 2011).
Another important example of SIRT1 in FH is the regulation of ribosomal DNA (rDNA) repression in the nucleolus. Due to its highly repetitive nature, the rDNA locus is a strong candidate for homologous recombination events, leading to unwanted chromosomal rearrangements. Nucleolar SIRT1, SUV39h1 and the H3K9me2-binding protein nucleomethylin are all components of the e-NoSc complex, which silences the rDNA locus by controlling ribosome biosynthesis under nutrient or energy scarcity (Murayama et al. 2008). Therefore, this complex provides a regulatory link between cellular energy balance and the epigenetic state of the rDNA locus (Murayama et al. 2008).
Constitutive Heterochromatin (CH)
Constitutive heterochromatin are always maintained as heterochromatin, play a structural role in the physical organization of the genome in the nucleus, contain few genes and are located primarily in pericentromeric regions and telomeres. By maintaining the structure of CH, Sirtuins play a crucial role in genomic stability. To date, only SIRT1 and SIRT6 have been directly linked to CH.
SIRT1 is the main Sirtuin involved. It has been related to pericentromeric heterochromatin (PCH) and telomere regulation (Vaquero et al. 2007). Indeed, loss of SIRT1 in mice has been associated with a loss of PCH foci and with derepression of the underlying γ-satellites (Bosch-Presegué et al. 2011). Involvement of SIRT1 in CH is directly related to its functional relationship with SUV39h1, a keystone of CH formation (Peters et al. 2001): loss of SIRT1 impairs SUV39h1-dependent H3K9me3 and HP1 localization in PCH (Vaquero et al. 2007). Given the limited amount or even absence of SIRT1 in PCH foci, one possible explanation for this functional loss is that SIRT1 might regulate PCH structure by preventing proteasomal degradation of SUV39H1. In vivo this increase in the levels of SUV39h1 results in its fast turnover in pericentromeric heterochromatin regions, which contributes to genome protection. Thus, stress leads to SIRT1-dependent upregulation of SUV39h1 in vivo, suggesting a direct link between the stress response and SUV39h1 dynamics in heterochromatin structure as a mechanism of genome stability (Bosch-Presegué et al. 2011).
The role of SIRT1 in telomeres is controversial. Some evidence suggests that SIRT1 inhibits telomerase activity by regulating the stability of the telomerase reverse transcriptase (TERT) catalytic subunit (Narala et al. 2008). In contrast, a battery of other findings suggests the opposite. For instance, Sirt1−/− cells exhibit reduced genome stability as well as telomeric aberrations that contribute to decreased cell growth (El Ramy et al. 2009). Furthermore, silencing of SIRT1 results in reduced expression of the shelterin complex components TERT and PTOP (Chen et al. 2011). Moreover, SIRT1 interacts with murine telomeric repeats, thereby attenuating the telomere shortening associated with ageing. Finally, SIRT1super mice, which contain an extra copy of the SIRT1 gene, show improved telomere maintenance owing to telomerase activation (Palacios et al. 2010).
SIRT6 also has been linked to telomere integrity maintenance, as it is critical for preventing telomere dysfunction and aberrant chromosomal end-to-end fusions. Deacetylation of H3K9Ac and H3K56Ac by SIRT6 in telomeres is required for efficient binding of helicase WRN to telomeres during the S-phase, to ensure efficient telomere replication and to prevent accumulation of structural abnormalities at telomeres (Michishita et al. 2008, 2009; Yang et al. 2009). Considering that H3K9Ac and H3K56Ac are also targets of SIRT1 and SIRT2, there might be some functional redundancy among Sirtuins in this context. Additionally, SIRT6 regulates telomere-proximal transgene silencing and has been linked to telomere position effect variegation (Tennen et al. 2011).
5.3.1.2 DNA Damage Repair: Signaling and Response
Another important role for Sirtuins is in DNA repair. Sirtuins participate in the signaling and repair of single-strand breaks (SSBs) and double-strand breaks (DSBs) at different levels. To date, the Sirtuins known to have the greatest involvement in DNA repair are SIRT1 and SIRT6.
DNA SSB
Sirt6−/− mice suffer from increased radiation sensitivity, chromosomal aberrations and impaired DNA repair. They also exhibit a premature aging phenotype associated with impaired base excision repair (BER), the main repair pathway for SSBs. However, a role for SIRT6 in BER remains unknown, because SIRT6 does not appear to interact or co-localize with BER factors at the damage sites (Mostoslavsky et al. 2006). Nonetheless, SIRT6 seems to indirectly regulate BER by modulating chromatin density and accessibility to DNA damage sites (Jia et al. 2012). Under oxidative stress conditions, SIRT6 is recruited to DNA damage sites, where it stimulates repair through direct interaction and ADP-ribosylation of PARP1, an enzyme involved in BER and DSB signaling. The role of SIRT6 as PARP1 activator might explain the deficiencies in BER and in genomic stability related to SIRT6 depletion (Mao et al. 2011).
SIRT1 has been linked to the nucleotide excision repair (NER) pathway for the repair of SSBs. Deacetylation of xeroderma pigmentosum complementation group A (XPA) by SIRT1 increases its interaction with RPA32, an essential event for the NER pathway (Fan and Luo 2010). SIRT1 also regulates the expression of XPC by reducing AKT-dependent nuclear localization of its transcriptional repressor (Ming et al. 2010).
DNA DSB
SIRT1, SIRT6 and, to a lesser extent, SIRT2, are involved in DSB repair at different levels. Upon γ-irradiation, SIRT1 plays a crucial role in the very early stages of DSB signaling, by modulating the formation of γ-H2AX, BRCA1, Rad51 and NBS1 foci (Wang et al. 2008). SIRT1-dependent deacetylation of NBS1, a regulatory subunit of the MRE11-Rad51-NBS (MRN) complex, modulates its activity as an intra-S phase checkpoint factor. Furthermore, relocalization of SIRT1 to DSBs depends on ATM-mediated signaling through H2AX phosphorylation (Oberdoerffer et al. 2008). SIRT1 also participates in DSB signaling through hMOF and TIP60, two members of the MYST family of histone acetyltransferases that are involved in cell growth (and growth arrest), apoptosis and DNA repair. Under normal conditions, SIRT1 binds to and deacetylates hMOF and TIP60, inhibiting their activities and promoting their ubiquitination-dependent degradation. However, DNA damage results in decreased binding of SIRT1 to both hMOF and TIP60 and activates DNA-damage signaling (Peng et al. 2012; Sun et al. 2009).
Interestingly, both SIRT1 and SIRT6 are directly involved in the two major DSB repair pathways: homologous recombination (HR) and non-homologous end joining (NHEJ). SIRT6 apparently plays an important role in HR, promoting DNA-end resection through interaction with, and deacetylation of, C-terminal binding protein interacting protein (CtIP) (Kaidi et al. 2010). CtIP is constitutively acetylated, but upon DNA damage, it is deacetylated by SIRT6 to promote resection together with Breast cancer 1 (BRCA1). Accordingly, SIRT6 loss results in reduced rates of HR and in sensitization of cells to DSB-inducing agents. Interestingly, a decrease in HR observed in replicative aging can be restored by SIRT6 overexpression. SIRT6 also interacts with factors of the NHEJ pathway, including DNA-PKcs and Ku70/80. SIRT6 depletion strongly diminishes recruitment of DNA repair factors in vitro and in vivo (Toiber et al. 2013). Accordantly, SirT6 is required for changes in chromatin structure surrounding DSBs, as it promotes a decrease in H3K9Ac levels to stabilize the association of DNA-PKcs to chromatin and to enable repair factors to access the DNA lesions (McCord et al. 2009). Additionally, deacetylation of H3K56 by SIRT6, and interaction of SIRT6 with the ATP-dependent chromatin remodeler SNF2H, are required for efficient DSB repair.
SIRT1 is linked to HR through its interaction and deacetylation of WRN helicase, a member of the RecQ family (Uhl et al. 2010). SIRT1 has been linked to NHEJ through deacetylation of the factor Ku70, which results in induction of Ku70-dependent DNA repair and inhibition of apoptosis through nuclear retention of the proapoptotic factor BAX (Cohen et al. 2004; Sawada et al. 2003).
Little is known about SIRT2 in DSB repair. SirT2 deacetylates H3K56Ac (Vempati et al. 2010) during S-phase and H4K16Ac during G2/M (Vaquero et al. 2006). Both marks are involved in DNA damage-repair response and are cell-cycle-dependent. Under normal conditions, H3K56Ac is spread throughout the nucleus; however, upon DNA damage, its levels increase and it concentrates in DNA damage foci, where it colocalizes with γ-H2AX, pATM, Chk2 and p53 (Vempati et al. 2010). Contrariwise, deacetylation of H4K16Ac at DNA damage sites is required for recruitment of 53BP1 to DSBs and for ensuring NHEJ repair (Hsiao and Mizzen 2013). SIRT2 is also linked to DNA repair through its ability to promote deposition of H4K20me1 during G2/M. Interestingly, establishment of H4K20me1 is also required for deposition of di- and tri- H4K20 methylation (H4K20me2/3) throughout the cell cycle. H4K20me1 levels indirectly affects DNA damage repair, since H4K20me2 is required for recruitment of 53BP1 to the DNA damage foci (Hartlerode et al. 2012; Serrano et al. 2013). However, some studies suggest that a portion of the H4K20me2 is not derived from the mitotic H4K20me1 and is deposited de novo in S-phase at DSB foci by the HMT MMSET (Pei et al. 2011).
5.3.1.3 Cell-Cycle Control
Regulation of cell-cycle progression is another mechanism for maintaining genome integrity. By regulating cell-cycle arrest, cells provide the DNA repair machinery with sufficient time to fix damage generated during the cycle or, in extreme cases, to activate apoptotic pathways. The Sirtuins most involved in cell-cycle control are SIRT2 and, to a lesser extent, SIRT1.
The link between SIRT2 and genome stability is well defined, as evidenced by the finding that Sirt2−/− mice exhibit major genome instability and are prone to develop tumors. Several studies support the role of SIRT2-deacetylase activity in the control of cell-cycle progression. SIRT2 is shuttled from the cytoplasm to the nucleus during the G2/M transition (Vaquero et al. 2006), where it is phosphorylated at S368 by CDK1. At the end of mitosis, CDC14A and CDC14B dephosphorylate SIRT2, thereby promoting its degradation and enabling mitotic exit (North and Verdin 2007). In fact, some evidence suggests that before being degraded, SIRT2 might also be involved in mitotic exit (Dryden et al. 2003). During the G2/M transition, SIRT2 controls genome stability by modulating chromatin condensation through deacetylation of H4K16Ac and regulation of H4K20me1 deposition. Thus, SIRT2 regulates the activity and the chromatin binding of the H4K20me1 HMT PR-SET7 and has a crucial role in spreading of H4K20me1 throughout the chromosome, thereby ensuring correct packing of chromatin during G2/M. Thus, SIRT2-dependent deacetylation of PR-SET7 increases its stability in chromatin and its catalytic activity towards H4K20. This regulation is crucial for the G2/M transition and is directly involved in G2/M checkpoint control. Under stress during G2/M, SirT2 binds strongly to PR-SET7, leading to an increased level of H4K20me1 and preventing cells from entering mitosis (Serrano et al. 2013). SirT2 is also related to the metaphase/anaphase checkpoint—probably via regulation of centrosome replication and modulation of Anaphase Promoting Complex/Cyclosome (APC/C) activity. Thus, SirT2 is partially localized in centrosomes (Kim et al. 2011), where it interacts with Aurora A, an essential protein for centrosome replication (Cowley et al. 2009). SirT2 deacetylates CDH1 and CDC20, two E3-ligases of the APC/C complex, which is essential for mitotic progression, which positively regulates its activity. After SirT2-dependent deacetylation, CDH1 and CDC20 bind APC/C complex, promoting Aurora-degradation, preventing abnormalities in centrosome replication and enabling mitotic exit (Kim et al. 2011). SIRT2 is also required for S-phase progression after replication stress. Stalled replication forks containing ss-DNA activate the replication stress response (RSR), which involves cell-cycle arrest, and stabilization and recovery of the stalled replication forks, to maintain genome stability. In this context, SIRT2 deacetylates and activates CDK9, a kinase required for the replication stress response. Accordingly, cells lacking either SIRT2 or CDK9 exhibit delayed S-phase progression after replication stress, due to impaired recovery from transient replication arrest (Zhang et al. 2013).
The role of SIRT1 in the cell cycle is related to the G1/S transition and to mitosis. In the first case, SIRT1 regulates the G1/S factors Retinoblastoma (Rb) and E2F1. Rb is a tumor suppressor that controls the G1/S transition by binding to E2F-responsive genes. SIRT1 deacetylates Rb and promotes its phosphorylation, thereby inhibiting Rb-dependent apoptosis and promoting cell proliferation (Wong and Weber 2007). Moreover, SIRT1 can also regulate the transcriptional repressive activity of the HDAC1-containing Rbp1 complex, thereby inhibiting its growth-arrest activity (Binda et al. 2008). In the second case, SIRT1 deacetylates histones and participates in loading of the Condensin I complex and H1 in chromatin, thereby facilitating chromosome condensation. SIRT1 depletion disrupts this loading and results in improper chromatin condensation and aberrant mitosis (Fatoba and Okorokov 2011).
5.3.2 Regulation of Gene Expression
As previously mentioned, an important function of Sirtuins in stress response is their role in regulating the stress-response expression programs of numerous transcription factors, including NF-κB, p53, HIF-1α, FOXOs, E2F1, PGC-1α and HSF1 (Fulco et al. 2003; Luo et al. 2001; Muth et al. 2001; Senawong et al. 2003; Takata and Ishikawa 2003; Vaziri et al. 2001). The Sirtuins most clearly involved in these processes are SIRT1, SIRT6 and, to a lesser extent, SIRT7.
Among the most important of the aforementioned transcription factors are the members of the FOXO family. FOXOs are involved in controlling the G1/S and G2/M checkpoints, and in inducing expression of genes involved in DNA-damage response, differentiation, glucose metabolism, and apoptosis (Huang and Tindall 2007). Under oxidative stress, Sirtuins target FOXO transcription factors, thereby dictating their subcellular localization, protein stability, and transcriptional activity (Brunet et al. 2004). Acetylation of FOXOs reduce their DNA binding and enhance their phosphorylation and inactivation. Deacetylation of FOXO by SIRT1 alters its interactions with E2F1 or p53, leading to apoptosis or to cell-cycle arrest, depending on the cellular metabolic state, environmental conditions, and tissue. Under oxidative stress, SIRT1 deacetylates FOXO3a, thereby increasing its capacity to promote cell-cycle arrest and preventing the cell from inducing apoptosis. Furthermore, SIRT1 enhances the cellular defense response to oxidative stress, by deacetylating and activating FOXO1 and FOXO4, inducing the expression of the cyclin/cdk inhibitor p27Kip1, manganese superoxide dismutase (MnSOD) and Growth arrest and DNA damage-inducible protein (GADD45) (Brunet et al. 2004; Daitoku et al. 2004; van der Horst and Burgering 2007). Analogously to SIRT1, SIRT2 deacetylates FOXO3a, enhancing its DNA binding and promoting expression of FOXO target genes, p27Kip1, MnSOD and the pro-apoptotic protein Bim. Consequently, SIRT2 promotes cell-cycle arrest, reduces cellular levels of ROS and, under severe stress, triggers apoptosis (Wang et al. 2007) (Fig. 5.2a).
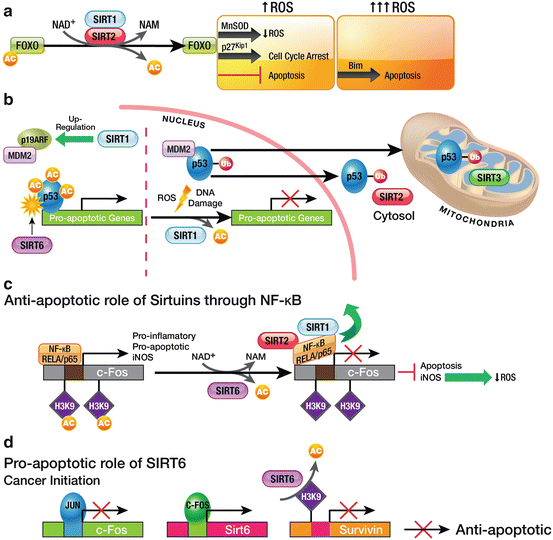
Fig. 5.2
Sirtuins in apoptotic pathways. (a) SIRT1 and SIRT2-dependent regulation of FOXO under oxidative stress. Under mild oxidative stress, SIRT1 and SIRT2 promote reduced ROS production and cell-cycle arrest, by activating the FOXO target genes MnSOD and p27Kip1, respectively. Under severe stress, this regulation triggers apoptosis. (b) Sirtuin-dependent regulation of p53. SIRT1 seems to regulate replicative life span by up-regulating p19ARF, consequently decreasing MDM2-dependent degradation of p53. Under stress, SIRT1 deacetylates p53, thereby promoting its monoubiquitination and its shuttling from the nucleus to the cytosol and mitochondria. Depending on the cellular state, SIRT1 either promotes or inhibits apoptosis through p53. SIRT6 activates p53 by mono[ADP-ribosyl]ating it. (c) Anti-apoptotic role of Sirtuins in pro-inflammatory pathways. SIRT6 is recruited by the NF-κB subunit RELA/p65 to pro-inflammatory genes, where it blocks NF-κB signaling by deacetylating H3K9Ac. In this context, SIRT1 and SIRT2 bind to NF-κB, removing it from chromatin and thereby contributing to the end of the pro-inflammatory signal. (d) Role of SIRT6 in oncogenesis: deregulation of the c-JUN pathway. SIRT6 represses expression of the anti-apoptotic gene survivin by deacetylating H3KAc in its promoter. Expression of Sirt6 is promoted by c-FOS, whose expression is repressed by c-JUN. During oncogenesis, c-JUN inhibits expression of c-fos and consequently, blocks repression of survivin, thereby leaving the cell to proliferate
Another important transcription factor is nuclear factor-κB (NF-κB), which is vital for regulating expression of certain genes involved in aging, proliferation and inflammation. Different subunits of the NF-κB family are acetylated and deacetylated at multiple sites, affecting its DNA-binding and transcriptional activity, and consequently, modulating the release of pro-inflammatory mediators. In this sense, SIRT6 plays an anti-inflammatory role, by regulating the NF-κB pathway and decreasing NF-κB-dependent apoptosis and senescence. Following activation of NF-κB by Tumor necrosis factor alpha (TNF-α), SirT6 interacts with the NF-κB subunit RELA/p65 and is recruited to promoters of a subset of NF-κB target genes. Subsequently, SIRT6 deacetylates H3K9Ac at those promoters and destabilizes RELA-promoter interaction, thereby contributing to NF-κB signal termination (Kawahara et al. 2009) (Fig. 5.2c). However, SIRT6 is not the only Sirtuin that modulates this pathway: SIRT1 and SIRT2 each interact with and deacetylate the RELA/p65 subunit, which results in NF-κB-mediated transcription inhibition (Rothgiesser et al. 2010; Oeckinghaus and Ghosh 2009). This effect in turn suppresses induction of nitric oxide synthase (iNOS) and nitrous oxide production, and might reduce the amount of cellular ROS (Lee et al. 2009). Additionally, treatment of cells with Resveratrol, a SIRT1 activator, correlates to a loss of expression of NF-κB-regulated genes and to sensitization of cells to TNFα-induced apoptosis. These findings suggest that SIRT1 activity, via NF-κB inhibition, augments apoptosis in response to TNFα (Yeung et al. 2004).
Interestingly, another anti-inflammatory role for SIRT6 has been described in Sirt6−/− mice. This role involves the transcription factors c-JUN and c-FOS, which are members of the AP-1 signal-transducing family and are implicated in cell-cycle progression, cell differentiation and apoptosis. c-FOS regulates SIRT6 expression. In turn, SIRT6 interacts with c-JUN and deacetylates H3K9Ac at promoters of pro-inflammatory genes including TNFα, MCP-1 and IL-6 (Xiao et al. 2012). Accordingly, SIRT6 represses expression of the anti-apoptotic protein survivin by H3K9Ac deacetylation. During liver tumor initiation, c-JUN regulates survival of initiated cancer cells through repression of c-fos transcription, which in turn downregulates SIRT6 expression, enabling expression of survivin and tumor initiation (Fig. 5.2d). In fact, overexpression of SIRT6 during liver tumor initiation is sufficient to decrease subsequent liver tumorigenesis (Min et al. 2012).
Stress affects more than just regulation of protein-coding genes: genotoxic stress, CR and other types of stress can also alter transcription of ribosomal RNA (rRNA). RNA-polymerase I (Pol-I) transcription has been shown to be repressed by nucleolar SIRT1, via deacetylation of its basal component TAFI68 (Ford et al. 2006). Interestingly, SIRT7 is a component of the Pol-I transcriptional machinery and enhances its transcriptional activity, performing an antagonistic role to that of SIRT1. SIRT7 depletion results in decreased association of Pol I with rDNA and consequently, in reduced Pol-I transcription. During mitosis, SIRT7 is phosphorylated and remains localized to the nucleolus until telophase, when it is activated (by dephosphorylation) to help rDNA expression resume after mitosis. SIRT7 has also been linked to repress transcription of a set of non-nucleolar genes via deacetylation of H3K18Ac at their promoters. SIRT7 deacetylates H3K18Ac at promoters of a network of genes linked to tumor suppression through its interaction with the cancer associated transcription factor ELK4. This SIRT7-associated function is related to the maintenance of an oncogenic phenotype in transformed cells, including anchorage-independent growth and escape from contact inhibition, which suggests an important role for SIRT7 as a tumorigenic factor (Barber et al. 2012). Another major category of genes targeted by SIRT7 is the ribosomal protein genes, which are transcriptional targets of the oncogenic protein MYC. In this context, SIRT7 plays an adaptive role in the Unfolded Protein Response (UPR) to suppress endoplasmic reticulum (ER) stress. During ER stress, cells must stop protein biosynthesis. Under these conditions, SIRT7 is up-regulated and is stabilized by MYC to the promoters of ribosomal protein genes, repressing its transcription and preventing the cell from initiating new protein biosynthesis. During hypoxia, cancer cells must decrease their energy expenditure and focus on survival. To this end, SIRT7 reduces expression of ribosomal protein genes, thereby enabling tumor progression (Shin et al. 2013).
5.3.3 Metabolic Energetic Mechanisms
Among the most important and conserved roles of Sirtuins is to modulate metabolic adaptation to different types of stress, as evidenced by the fact that three Sirtuins (SIRT3, SIRT4 and SIRT5) are localized to the mitochondria, where they control mitochondrial energy production, substrate oxidation and apoptosis (Onyango et al. 2002; Jeong et al. 2013).
Sirtuins have been linked to CR, which extends lifespan and reduces ROS production by shifting cell metabolism from glycolysis to fatty acid oxidation, amino acid metabolism and ketogenesis (Fig. 5.3). The mitochondrial Sirtuins, together with SIRT1 and SIRT6, apparently coordinate this metabolic shift—a function that would intertwine them with CR, aging and carcinogenesis (Koubova and Guarente 2003; Qiu et al. 2010). For example, under CR, SIRT3 is up-regulated (Han and Someya 2013) and in turn, deacetylates acetyl-CoA synthetase 2 (AceCS2). AceCS2 is expressed in muscle and converts acetate into acetyl-CoA under ketogenic conditions such as CR, thereby enabling muscle to use acetate as a carbon source (Schwer et al. 2006).
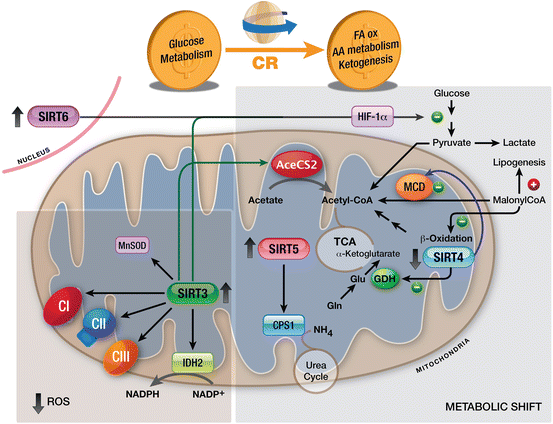
Fig. 5.3
Mitochondrial Sirtuins and SIRT6 during CR. CR shifts cell metabolism from glycolysis to FA oxidation, AA metabolism and ketogenesis. Increased activity of SIRT3 promotes reduced production of ROS by activating IDH2, the electron transport chain and MnSOD. Moreover, SIRT3, SIRT4, SIRT5 and SIRT6 drive the metabolic shift triggered by CR. Abbreviations: AA amino acid, CR calorie restriction, FA fatty acid, TCA tricarboxylic acid
Among the functional connections between Sirtuins and metabolism is regulation of mitochondrial targets by acetylation/deacetylation. Most mitochondrial proteins are acetylated, including the major regulators of mitochondrial metabolism, and all three mitochondrial Sirtuins have been shown to deacetylate mitochondrial targets (Lombard et al. 2007; Haigis et al. 2012; Nakagawa and Guarente 2009). Among mitochondrial deacetylases, SIRT3 seems to predominate, as indicated by the finding that mitochondrial proteins are massively acetylated in Sirt3−/− mice. However, a small pool of SIRT3 can also be found in the nucleus under normal conditions, where it deacetylates H4K16Ac and H4K56Ac, which are essential marks in DNA damage response and in chromatin condensation (Scher et al. 2007; Vempati et al. 2010).
SIRT3 also seems to have an important role in ROS detoxification at different levels. Firstly, activation of SIRT3 might help increase mitochondrial respiration and ATP production, as indicated by the finding that SIRT3 deacetylates and activates complex I-III of the electron transport chain, thereby ensuring proper reduction of oxygen and avoiding ROS production (Cimen et al. 2010). Secondly, under CR, SIRT3 activates isocitrate dehydrogenase 2 (IDH2), an enzyme responsible for the conversion of NADP+ to NADPH in mitochondria (Someya et al. 2010). Thirdly, by deacetylating the transcription factor FOXO3a, SIRT3 regulates the expression of manganese superoxide dismutase (MnSOD), an enzyme responsible for superoxide detoxification (Kim et al. 2010). Additionally, SIRT3 deacetylates MnSODK122, thereby increasing its enzymatic detoxification activity (Tao et al. 2010). Altogether, regulation of the electron transport chain, and of MnSOD activity and expression, by SIRT3 maintains the cellular balance of ROS. Accordingly, upon exposure to ionizing radiation, Sirt3−/− MEFs exhibit increased levels of superoxide, decreased stress-induced apoptosis, infrequent contact inhibition and increased chromosome instability (Sundaresan et al. 2009; Kim et al. 2010).
Evidence suggests that SIRT3 and SIRT6 each act as tumor suppressor by regulating cancer cell metabolism. Loss of SIRT3/SIRT6-dependent metabolic regulation has been directly associated with a metabolic switch to anaerobic glycolysis, known as the Warburg effect, which is considered a hallmark of cancer cells (Kim et al. 2010; Zhong et al. 2010). This switch leads to a situation very similar to hypoxia. The major regulatory factor of hypoxia is HIF-1, which comprises a heterodimer of HIF-1α and HIF-1β. During normoxia, HIF-1α is hydroxylated by a family of oxygen-dependent prolyl hydroxylases (PHD1-3), flagging HIF-1α for ubiquitination and subsequent proteasomal degradation. However, under hypoxia, HIF-1α is stabilized and promotes expression of certain HIF-1α dependent pro-proliferative and pro-survival genes. Hypoxia also triggers an increase in ROS production, which inhibits PHD and consequently, stabilizes HIF-1α. Interestingly, the mechanism by which SIRT3 regulates glycolysis involves HIF-1α and its target genes: SIRT3 maintains ROS equilibrium, thereby preventing inactivation of PHD and keeping HIF-1α levels low (Bell et al. 2011). Likewise, SIRT6 controls glucose homeostasis by inhibiting several glycolytic genes (by deacetylating H3K9Ac), including many that are targets of HIF-1α. Accordingly, Sirt6−/− mice present hyperacetylation of H3K9 at these promoters and increased gene expression, which results in increased glucose uptake and glycolysis, boosting cell growth and proliferation (Zhong et al. 2010). SIRT6 has also been found to corepress MYC transcriptional activity towards ribosomal genes. Therefore, SIRT6 acts as an important tumor suppressor, modulating glycolysis and inhibiting ribosome biosynthesis, by corepressing HIF-1α and MYC transcriptional activity.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


