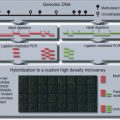
Figure 21.22.1 MAPit overview for mapping m5CG and chromatin accessibility in mammalian nuclei. Nuclei are isolated from cultured cells of interest grown under desired experimental conditions. Isolated nuclei are then probed with M.CviPI, a DNA methyltransferase that methylates cytosines at accessible GC sites, i.e., nucleosome-free or unbound by non-histone proteins. After purification of DNA, cytosines will either be unmethylated (unfilled symbols), modified at CG by endogenous DNMTs (m5CG; black-filled circles), or modified at GC by M.CviPI probe (G-m5C; red-filled inverted triangles). Purified DNA is then subjected to chemical conversion by bisulfite ion in which unmethylated C in denatured DNA is deaminated to U, whereas methylated C is not. During subsequent PCR of bisulfite-treated DNA, U is converted through replicative complementarity to T and m5C to C. Sequencing after cloning individual DNA molecules from the bulk PCR product provides a single-molecule readout of the methylation status at C in each CG and GC site. This output is representative of the combined endogenous methylation and chromatin accessibility at a single locus within the original nuclei. Sequence alignment and visual representation of methylation status are efficiently obtained computationally, e.g., with MethylViewer (Pardo et al., 2010). In the shown view in the bottom panel, each horizontal line indicates a single sequenced DNA molecule decorated with symbols representing the methylation status of each CG, GC, and overlapping CGC as defined (key at bottom right).
MAPit can be performed on organisms that lack detectable DNA methylation, such as the budding yeast Saccharomyces cerevisiae (Proffitt et al., 1984). Readers interested in MAPit probing of budding yeast chromatin are urged to consult any of several previously published detailed protocols (Jessen et al., 2004; Hoose and Kladde, 2006; Kilgore et al., 2007; Pardo et al., 2009). In principle, a DNMT probe can be used to modify cytosine in any sequence context on one of several positions. This is because bisulfite sequencing detects m5C as well as methyl-N4-C (mN4C) and hydroxymethyl-5-C (hm5C) (Vilkaitis and Klimasauskas, 1999; Kriaucionis and Heintz, 2009; Tahiliani et al., 2009; Huang et al., 2010). Therefore, in organisms lacking detectable DNA methylation, the choice of DNMT need only consider the frequency and distribution of target sites on the analyzed strand of the locus of interest. However, C-modifying DNMTs with short recognition specificities, such as the C-5 DNMTs M.SssI (CG; Renbaum et al., 1990) and M.CviPI (GC; Xu et al., 1998a), are the most useful probes for MAPit analysis because they provide the highest footprinting resolution. An additional consideration for probe choice is that most genomic regions in vertebrate cells contain some m5CG, limiting the usefulness of the CG-modification enzyme M.SssI (Fatemi et al., 2005; Gal-Yam et al., 2006). For this reason, the authors previously identified, cloned, overexpressed, and characterized the GC probe M.CviPI (Xu et al., 1998a), which is now commercially available.
Materials
Trypsin-EDTA solution (see recipe), 37°C
Mammalian cell lines cultured under appropriate experimental conditions in tissue culture plates or flasks
Cell growth medium (store up to 4 months at 4°C), 37°C
Phosphate buffered saline (PBS; APPENDIX 2), ice cold
0.4% (w/v) trypan blue solution (store indefinitely at room temperature)
80 U/µl M.CviPI fused to maltose binding protein (MBP, New England Biolabs) or fused to glutathione-S-transferase (GST, Zymo Research); store in 20-µl aliquots up to 1 year at −20°C in non-frost-free freezer; see recipe for dilutions)
DNMT dilution buffer (see recipe), ice cold
DNMT storage buffer (see recipe), ice cold
Cell resuspension buffer (see recipe), ice cold
Cell lysis buffer (see recipe), ice cold
Methylation buffer (see recipe)
Methylation stop buffer (see recipe), room temperature
20 mg/ml proteinase K (store up to 4 months at −20°C in non-frost-free freezer)
Phenol/chloroform solution (see recipe)
10.0 M ammonium acetate, pH 8.0 (APPENDIX 2)
Absolute and 70% (v/v) ethanol (see recipe; store indefinitely at room temperature)
0.1× TE buffer (see recipe)
Refrigerated microcentrifuge
Hemacytometer or automated cell-counting device
Light microscope
1.7-ml microcentrifuge tubes
37° and 50°C water baths
Additional reagents and equipment for bisulfite sequencing (UNIT 7.9)
NOTE: Reagents should be prepared in sterile disposable labware. Use only distilled water in all steps and solutions. Nuclei isolation and methylation buffers should be freshly prepared on the day of the experiment. DTT, PMSF, and SAM should be added to solutions immediately before use to avoid oxidation or hydrolysis. M.CviPI activity is strongly dependent on fresh addition of DTT.
Harvest cells
1. Add an appropriate volume of 37°C trypsin-EDTA solution to remove cells from tissue culture plates or flasks (1 ml trypsin-EDTA per 10-cm culture dish). Incubate cells at room temperature until they detach from the growth surface.
Cell lines and growing conditions will vary according to the question being addressed and discretion of the researcher. Cells should be cultured using standard tissue culture techniques under desired experimental conditions until at least 1.5 × 106 cells per experimental sample (e.g., DNMT dose) are obtained. A refrigerated centrifuge and microcentrifuge or one in a cold room is recommended for isolation of nuclei.
The time needed for cell detachment varies from one cell line to another (~2 to 10 min), and can be determined by visualization with a light microscope. Alternatively, cells can be harvested by adding ice-cold PBS directly to plates and scraping into 50-ml conical tubes on ice. Skip this step and step 2 if cells have been grown in suspension.
2. Add cell growth medium pre-warmed to 37°C (three times the volume of trypsinization solution used in step 1) to terminate trypsinization.
Trypsin activity is inhibited by the protease inhibitor alpha-1-antitrypsin found in fetal bovine serum.
3. Centrifuge 5 min at 1000 × g, 4°C, to pellet cells. Carefully aspirate supernatant.
4. Add 5 ml ice-cold PBS. Pipet up and down gently and briefly to resuspend cell pellet and wash cells.
5. Centrifuge for 5 min at 1,000 × g at 4°C to pellet cells. Aspirate supernatant.
6. Resuspend cells with ice-cold PBS to an approximate concentration of 106 cells/ml and keep cells on ice.
This is equivalent to resuspending HeLa cells from a 10-cm plate at 90% confluence into 3 ml of PBS.
7. Mix a 20-µl cell suspension aliquot with 20 µl of 0.4% (w/v) trypan blue solution. Pipet cells up and down several times to disperse and make a homogeneous cell suspension.
8. Count the number of live cells that exclude trypan blue either manually with a hemacytometer or by using an automated cell-counting device.
9. Dispense 1.1 × 106 cells per experimental sample into pre-labeled 1.7-ml microcentrifuge tubes on ice.
Each DNMT probing reaction requires 106 cells. Starting with 1.1 × 106 cells per reaction (one reaction is one DNMT dose) allows for some loss during preparation of nuclei. It is recommended to set up an untreated sample (0 U DNMT) and two concentrations of M.CviPI, therefore requiring 3.3 × 106 cells per experimental condition. In the authors’ experience, 30 and 100 U M.CviPI are good starting doses for either the M.CviPI-MBP or M.CviPI-GST reagents. Using two different concentrations of enzyme, while not essential, allows one to assess different degrees of chromatin accessibility and the extent of saturation of methylation at each GC site by exogenously added M.CviPI. The untreated sample (0 U DNMT) serves as a background control to monitor non-conversion of C in GC sites by bisulfite and/or sequencing errors. The untreated sample also shows the level of endogenous CG methylation in the sample before probing, which should be taken into account when inferring whether GCG sites were likely methylated by endogenous DNMTs or exogenously added by the DNMT probe.
10. Microcentrifuge 5 min at 1000 × g, 4°C, to pellet cells.
11. Add 200 µl ice-cold cell resuspension buffer per 1.1 × 106 cells (i.e., add 600 µl, if 0, 30, and 100 U M.CviPI are used). Resuspend pellet by tapping tube gently.
Isolating all nuclei for each experimental condition together in a single tube and dispensing into separate tubes in step 17 ensures that the only variable will be the DNMT concentration.
12. Centrifuge 5 min at 1000 × g, 4°C, to pellet cells. Decant supernatant.
Isolate mammalian nuclei
13. Resuspend cell pellet in 38.5 µl of ice-cold cell lysis buffer per 1.1 × 106 cells (i.e., add 115.5 µl if 0, 30, and 100 U M.CviPI are used). Incubate 10 min at 4°C to lyse cells.
Inclusion of the non-ionic detergent Nonidet P-40 in cell lysis buffer allows for cell membrane lysis while maintaining nuclear integrity. Nonidet P-40 concentration and lysis time may need to be optimized for different cell types to obtain complete cell lysis without disrupting the integrity of the nuclear envelope. To preserve nuclear structural integrity and native protein-DNA interactions, all steps for nuclei preparation should be performed at 4°C. Nuclei should be handled carefully as they are prone to lysis. Avoid pipetting of nuclei until step 17; instead, resuspend by gentle tapping of the tube with a finger.
14. Prepare ice-cold methylation buffer while cells are undergoing lysis by mixing on ice the following:
60.5 µl ice-cold cell resuspension buffer
0.55 µl freshly-thawed 32 mM SAM.
These volumes are per each sample containing ~106 nuclei. Prepare enough extra solution to account for pipetting errors. The methylation buffer contains 290 µM SAM, which will be diluted to a final concentration of 160 µM in the methylation reactions in step 20.
15. Check the efficiency of cell lysis and structural integrity of the nuclei. Stain a 2-µl aliquot of nuclei solution by adding 2 µl of 0.4% (w/v) trypan blue solution in a separate tube. Mix by gently tapping the tube and immediately examine nuclei by light microscopy.
Nuclei should stain blue as well as appear round and granular with no attached cytoplasmic debris. Nuclei may swell slightly during isolation and subsequent manipulations.
16. Add 61 µl ice-cold methylation buffer per 106 nuclei (i.e., add 183 µl if 0, 30, and 100 U M.CviPI are used) to dilute Nonidet P-40 concentration. Mix by gently tapping the tube.
Dilution of Nonidet P-40 detergent to 0.07% (v/v) in this step helps maintain nuclear integrity and enable full DNMT activity.
17. Dispense 90 µl of nuclei resuspension containing 106 nuclei into 1.7-ml microcentrifuge tubes pre-labeled with each unit concentration of M.CviPI being used.
Probe nuclear chromatin by methylation with exogenous M.CviPI
18. On ice, freshly prepare M.CviPI dilutions for the methylation reaction.
For the 30 and 100 U M.CviPI samples, appropriate volumes of 3 U/µl and 10 U/µl M.CviPI solutions, respectively, are needed. Best results are achieved by serial dilution of the concentrated M.CviPI stock with ice-cold, freshly made DNMT dilution buffer. This ensures that all samples are subjected to identical conditions in parallel (i.e., salt and 160 µM SAM), with the DNMT concentration being the only variable.
19. Pre-warm the nuclei dispensed in each tube by incubating 2 min at 37°C. At the same time, pre-warm to 50°C a sufficient volume of 2× methylation stop buffer (100 µl per methylation reaction plus some extra to allow for pipetting errors).
20. Stagger M.CviPI addition to each tube by 30 sec, add 10 µl of the corresponding M.CviPI dilution to each pre-warmed sample, i.e., add 10 µl of DNMT dilution buffer to the 0 U sample, 10 µl of 3 U/µl M.CviPI dilution to the 30 U sample, and 10 µl of the 10 U/µl M.CviPI dilution to the 100 U sample. Pipet up and down gently to mix and methylate for 15 min at 37°C.
Staggered addition of enzyme and respective staggered termination of methylation in step 21 ensure that the incubation time with the chromatin-probing enzyme is held constant. Parameters used during the chromatin probing reaction can be changed according to the requirements of the experiment. It is recommended to perform a pilot experiment under the conditions described here. Time and enzyme concentration can be adjusted accordingly (see Commentary).
21. Terminate each methylation reaction by adding 100 µl of 2× methylation stop buffer pre-warmed to 50°C, corresponding to the staggering scheme used in step 20. Vortex each sample immediately.
22. Add 1 µl of 20 mg/ml proteinase K to a final concentration of 100 µg/ml. Mix by inverting tubes and incubate overnight at 50°C.
Complete removal of protein from the DNA is necessary to achieve complete denaturation and hence bisulfite conversion (Warnecke et al., 2002). Incubation with proteinase K for at least 16 hr is required.
Purify mammalian genomic DNA
23. Extract proteins from the genomic DNA solution from step 22 by adding 200 µl (an equal volume) of phenol/chloroform solution (UNIT 2.3). Vortex vigorously for 30 sec at room temperature to obtain a homogeneous suspension.
24. Separate the aqueous and organic phases by centrifuging 5 min at 14,000 × g, room temperature, in a microcentrifuge. Transfer the aqueous (top) phase to a new 1.7-ml microcentrifuge tube carefully avoiding transfer of both denatured protein and SDS (the white material located at the phase interface).
25. Add 1/4 vol of 10.0 M ammonium acetate (i.e., 2.5 M final), and vortex briefly to mix.
26. Add 2.5 vol of absolute ethanol, mix thoroughly by gentle inversion.
At this point, samples can be stored indefinitely at −20°C. Overnight incubation at −20°C increases recovery of low concentrations of nucleic acid.
27. Centrifuge 5 min at 14,000 × g, room temperature, in a microcentrifuge to pellet the nucleic acid.
28. Draw off supernatant carefully so as not to dislodge the nucleic acid pellet.
29. Add 0.4 ml of 70% (v/v) ethanol. Vortex briefly to wash nucleic acid pellet.
30. Centrifuge 5 min at 14,000 × g, room temperature, in a microcentrifuge to pellet nucleic acid.
31. Carefully draw off supernatant with a pipet without disturbing the pellet and air dry pellet for ~10 min.
32. Resuspend genomic DNA in 50 µl of 0.1× TE buffer.
Genomic DNA usually requires overnight incubation at 4°C to solubilize completely. Removal of RNA prior to bisulfite sequencing is not necessary. Samples can be stored for many months at 4°C or indefinitely at −20°C.
Perform bisulfite sequencing of mammalian DNA
33. Approximately 5 to 15 µg DNA are recovered from each reaction containing 10
6 nuclei. Bisulfite sequencing, including bisulfite conversion of purified DNA, PCR amplification of sequences of interest, cloning individual molecules from the PCR product, and sequencing cloned molecules, is performed as described in
UNIT 7.9. Once clones of individual molecules have been sequenced, the data are analyzed by MethylViewer (
http://dna.leeds.ac.uk/methylviewer/) (Pardo et al.,
2010). This computer program can concurrently score the methylation status of up to four user-defined sites either directly from *.ab1 sequencing files or from a FASTA file of sequences aligned with another program. For MAPit analysis of mammalian chromatin with M.CviPI, MethylViewer is used to concurrently score methylation at CG and GC sites along each sequenced molecule, and export publication-quality images. Other features, such as verification of bisulfite conversion efficiency at non-CG and non-GC sequences, can also be obtained. Occasional sequences with conversion efficiencies of <97% are typically omitted from further analyses, but this is up to the discretion of the investigator.
SUPPORT PROTOCOL
VERIFICATION OF METHYLATION OF DNA BY M.CviPI
When using a new enzyme preparation, one should determine activity before investing time in sequencing and analysis of MAPit data. It is possible to methylate purified plasmids and test with various restriction enzymes. However, higher enzymatic activity is needed to methylate chromatin. It is convenient to assay activity by methylation of nuclear chromatin, using the actual experimental samples. To confirm that the DNMT used was active, one of two methods may be used to screen for GC methylation, either quantitative methylation-sensitive restriction enzyme digestion (qMSRE) or methylation-specific PCR (MSP).
Quantitative methylation-sensitive restriction enzyme digestion (qMSRE)
For qMSRE, 20 to 250 ng of purified genomic DNA (not bisulfite treated) is subject to digestion with the methylation-sensitive restriction (R) enzyme R.HaeIII. This enzyme digests unmethylated GGCC sites but not GG-m5CC sites. A parallel “mock” reaction containing all reaction components except R.HaeIII (replaced with glycerol) is included for each sample. DNA from the R.HaeIII-digested or mock reaction is then amplified by real-time PCR with primers flanking a known open region containing a HaeIII site, such as the human GAPDH promoter (primers TACTAGCGGTTTTACGGGCG and TCGAACAGGAGGAGCAGAGAGCGA). Results are normalized to each sample’s mock digestion control and quantified by real-time PCR using the ΔΔCT method to determine the levels of protection from R.HaeIII digestion achieved by each dose of DNMT.
Methylation-specific PCR (MSP)
For MSP, 20 ng of bisulfite-treated DNA is amplified with two sets of primers that target human long interspersed nuclear elements (LINE-1). One primer pair amplifies GC unmethylated or “U” LINE (primer sequences AGGtATTGttTtAttTGGGAAGtGt and CCTTACAaTTTaATCTCAaACTACTaTA) and the second pair amplifies GC-methylated or “M” LINE (primers CATTGCtTtAttTGGGAAGCGC and CTTGCAaTTTaATCTCAaACTGCTaTG) sequences. Lower case letters in each primer indicate transitions either from C to t or from G to a, while underlining denotes the location of GC sites in the original, bisulfite-untreated sequences. Therefore, both the U and M primer pairs specifically amplify bisulfite-converted sequences that are, respectively, unmethylated (GC converted to Gt) and methylated (G-m5C converted to GC). The product of each PCR reaction is visualized on an agarose gel: the M product will be more abundant than the U product if the DNMT was active.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2; for suppliers, see APPENDIX 4.
Cell lysis buffer
Cell resuspension buffer (see recipe)
0.19% (v/v) Nonidet P-40
Prepare fresh and keep on ice
Cell resuspension buffer
20 mM HEPES, pH 7.5
70 mM NaCl (APPENDIX 2)
0.25 mM EDTA, pH 8.0 (APPENDIX 2)
0.5 mM EGTA, pH 8.0
0.5% (v/v) glycerol
10 mM DTT (APPENDIX 2; always add fresh immediately before use, store in single-use 400-µl aliquots at −20°C)
0.25 mM phenylmethylsulfonyl fluoride (PMSF; always add fresh immediately before use, store dissolved in absolute ethanol up to 6 months at −20°C)
Prepare fresh and keep on ice
DNMT dilution buffer (M.CviPI and M.SssI)
Dilute M.CviPI storage buffer by eight-fold (i.e., 1:7) with methylation buffer. Prepare fresh and keep on ice.
DNMT storage buffer (M.CviPI and M.SssI)