Sideroblastic anemias (SAs) may be acquired or congenital and share the features of disrupted utilization of iron in the erythroblast, ineffective erythropoiesis, and variable systemic iron overload. Congenital forms can have associated syndromic features or be nonsyndromic, and many of them have mutations in genes encoding proteins involved in heme biosynthesis, iron-sulfur cluster biogenesis, or mitochondrial protein synthesis. The mechanism(s) for the acquired clonal SA is undefined and is under intense study. Precise diagnosis of these disorders rests on careful clinical and laboratory evaluation, including molecular analysis. Supportive treatments usually provide for a favorable prognosis and often for normal survival.
Key points
- •
Sideroblastic anemias are diverse metabolic diseases of the erythroid cell.
- •
The presence of bone marrow ring sideroblasts is the diagnostic feature of all sideroblastic anemias.
- •
The disease course can usually be predicted when the underlying cause is identified.
- •
Systemic iron overload that occurs in the common sideroblastic anemias is mediated by ineffective erythropoiesis and leads to morbidity and reduced survival if untreated.
General overview
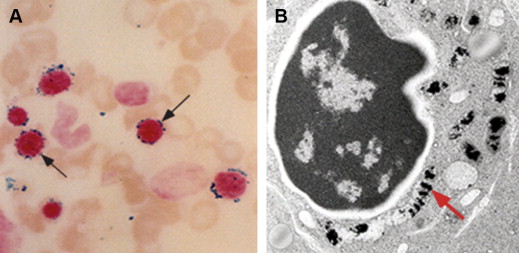
When first defined 50 years ago, sideroblastic anemia (SA) was already recognized to occur in heterogeneous settings, including as familial or acquired disease. The spectrum of SA has since become considerably expanded with respect to distinct clinical phenotypes as well as discrete causes. The singular feature that typifies all forms of SA and is required for initial diagnosis is the presence of telltale ring sideroblasts in the bone marrow aspirate smear. These erythroblasts contain numerous coarse Prussian blue–positive granules, characteristically appearing in a perinuclear distribution ( Fig. 1 A), which represent pathologic deposits of iron in mitochondria (see Fig. 1 B).
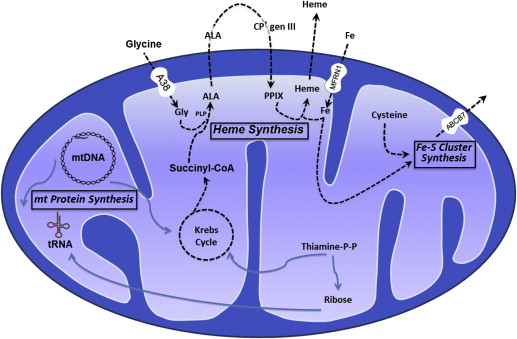
The magnitude of the quantity of iron used by the erythron to make hemoglobin is well appreciated. Hemoglobin contains nearly 80% of the body heme and therein more than two-thirds of the body iron. Heme is assembled from iron and protoporphyrin IX in the mitochondrion by ferrochelatase (FECH). However, iron imported into mitochondria is also required to generate iron-sulfur (Fe-S) clusters, which themselves participate as essential prosthetic groups in proteins regulating cellular iron uptake, heme synthesis, and iron storage, including iron regulatory protein 1 (IRP1) and FECH itself.
General concepts regarding the pathogenesis of the SAs have emerged from discoveries of the genetic defects underlying various congenital SAs (CSAs). Based on our current knowledge, the unique pathology can be primarily linked to abnormalities in the heme biosynthesis and Fe-S biogenesis pathways as well as to defects in the translation of mitochondrially encoded proteins ( Fig. 2 ). Nevertheless, in most instances, our understanding of the downstream events that lead to ring sideroblast formation and anemia is severely lacking. Importantly, body iron metabolism is also strikingly altered in the common SAs, in that the associated ineffective erythropoiesis elicits a systemic iron overload state, which often influences clinical features as well as prognosis, as discussed later.
Diagnosis
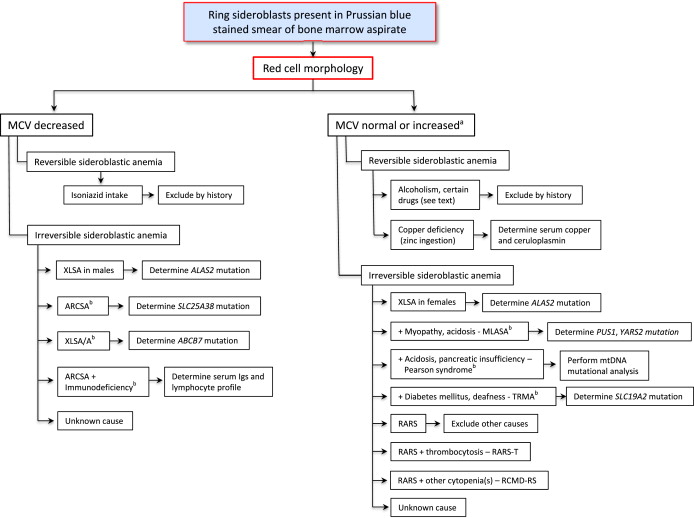
The SAs can be divided into CSA and acquired forms. Although they are inherited, the CSAs may not present at birth, and sometimes may not be recognized until adulthood. Because of certain distinctive treatments and considerably different prognoses, it is particularly important to distinguish later-onset CSA from an acquired SA. As summarized later, the recognition of the specific diagnosis relies on characteristics of the anemia (eg, microcytic, normocytic, or macrocytic), the age of clinical onset, associated syndromic features, and, increasingly, molecular genetic diagnostics ( Table 1 ).
| SA Class | Mutations Identified | Inheritance | Clinical Onset | Severity of Anemia | Mean Corpuscular Volume | Red Blood Cell Protoporphyrin | Associated Abnormalities | |
|---|---|---|---|---|---|---|---|---|
| Gene | Protein | |||||||
| CSA | ||||||||
| Nonsyndromic CSA | ||||||||
| X-linked (XLSA) | ALAS2 | ALAS2 | X-linked | Variable | Mild to severe | ↓ c | N/↓ | Iron overload |
| SLC25A38 deficiency | SLC25A38 | mt transporter SLC25A38 | AR | Childhood | Severe | ↓ | N/↓ | Iron overload |
| Glutaredoxin 5 deficiency a | GLRX5 | Glutaredoxin 5 | AR | Adulthood | Mild to severe | ↓ | Not reported | Iron overload |
| Erythropoietic protoporphyria (EPP) b | FECH | FECH | AR | Usually childhood | Mild | ↓ | Marked ↑ | Acute photosensitivity |
| Syndromic CSA | ||||||||
| X-linked with ataxia (XLSA/A) | ABCB7 | mt transporter ABCB7 | X-linked | Childhood | Mild to moderate | ↓ | ↑ | Incoordination, delayed motor development |
| SA, B-cell immunodeficiency, fevers and developmental delay (SIFD) | Unknown | Unknown | AR | Infancy | Severe | ↓ | Not reported | Iron overload, CNS derangements, deafness, cardiomyopathy |
| Pearson marrow-pancreas syndrome | mtDNA | Mitochondrial tRNAs | Sporadic/maternal | Early childhood | Severe | ↑ | ↑ | Metabolic acidosis, exocrine pancreatic insufficiency, hepatic/renal failure |
| Myopathy, lactic acidosis, and SA (MLASA) | PUS1/YARS2 | Pseudouridine synthase 1/mt tyrosyl tRNA synthetase | AR | Childhood | Mild to severe | N/↑ | Not reported | Myopathy, lactic acidosis/cardiomyopathy ± |
| Thiamine-responsive megaloblastic anemia (TRMA) | SLC19A2 | Thiamine transporter | AR | Childhood | Severe | ↑ | N/↑ | Non-type I diabetes, sensorineural deafness |
| Syndromic/nonsyndromic SA of unknown cause | Unknown | Unknown | Varied | Variable | Variable | Variable | Variable | ± diverse |
| Acquired SA | ||||||||
| Clonal/neoplastic | ||||||||
| RARS | SF3B1 | Core component of RNA spliceosome | — | Adulthood | Mild to severe | N/↑ | ↑ | Iron overload |
| RARS-T | Moderate | N/↑ | Not reported | Thrombocytosis | ||||
| RCMD-RS | Mild to severe | N/↑ | Not reported | Cytopenias | ||||
| Metabolic | ||||||||
| Alcoholism | — | — | — | Adulthood | Variable | N/↑ | Variable | Nutritional/toxic effects of alcohol |
| Drug-induced | — | — | — | Adulthood | Variable | Variable | Variable | None |
| Copper deficiency (zinc toxicity) | — | — | — | Any age | Variable | N/↑ | ↑ | Neurologic deficits, neutropenia |
| Hypothermia | — | — | — | 3 adults reported | Mild to moderate | N/↑ | Not reported | CNS features |
a Described in only 1 patient.
b Ring sideroblasts shown in 10 cases.
Nonsyndromic CSAs
X-linked SA (XLSA) is the most common CSA, constituting about 40% of cases. It is often mild or asymptomatic and may be discovered only in young adulthood or even in later life; occasional severe cases have presented in infancy or childhood. Rarely is it associated with an in utero (often lethal) phenotype. Not uncommonly, it is seen in women as a result of acquired skewed X chromosome inactivation in hematopoietic tissue, which occurs with advancing age. Skewed constitutional X chromosome inactivation to account for disease expression in females in childhood is exceptional.
Mutations in ALAS2, the first and rate-controlling enzyme of heme synthesis, cause XLSA. More than 80 distinct mutations have been identified in more than 120 unrelated kindreds or probands. About one-fifth of the mutations have occurred in more than 1 family, and nearly one-third of probands are female. Most of the mutations are missense alleles. Rare stop or frame shift alleles are viable only in females. Recently, several mutations affecting a GATA transcription factor binding site in intron 1 of the ALAS2 gene were discovered. These heterogeneous molecular defects predict diverse effects on the function of the ALAS2 enzyme and in turn variable severity of anemia.
An autosomal recessive CSA caused by mutations in SLC25A38, accounting for about 15% of CSAs, presents at birth or in early childhood as a severe, transfusion-dependent microcytic hypochromic anemia. The genetic defect involves the erythroid-specific mitochondrial inner membrane carrier protein SLC25A38, predicted to be an amino acid transporter required to import the ALAS2 substrate glycine, or possibly exchanging glycine for ALA, across the inner mitochondrial membrane in the initial steps of heme synthesis. Among 31 patients so far reported, mutations are heterogeneous and include missense as well as nonsense and splicing errors, with one-fourth of them being recurrent in unrelated families. Most mutations seem to be severe or complete loss-of-function mutations. Because of its autosomal location, this anemia is more likely seen in children of consanguineous parentage.
A single case of a microcytic hypochromic transfusion-dependent SA presenting in midlife has been attributed to a homozygous splice site mutation in the glutaredoxin 5 ( GLRX5 ) gene. In this instructive case, it could be shown that GLRX5 deficiency severely impairs Fe-S cluster biogenesis, which reduces ALAS2 translation through increased iron-responsive element binding activity of IRP1 lacking the Fe-S cluster, as well as the integrity of FECH deprived of its Fe-S cluster.
Ring sideroblasts have been documented in only 10 patients with erythropoietic protoporphyria (EPP), a disorder characterized by marked deficiency of FECH. Mild anemia is observed in most patients, but it is uncertain that EPP consistently results in a CSA marrow phenotype.
The clinical laboratory clue for each of the nonsyndromic CSAs is erythrocyte microcytosis and hypochromia (reduced mean corpuscular volume [MCV] and mean corpuscular hemoglobin), along with an increased red cell distribution width (RDW) and absence of evidence for common causes of microcytic anemia such as iron deficiency and thalassemia. An important exception to this characteristic is that most females expressing XLSA have a normal or increased MCV. In severely affected females, erythroid cells with a nonfunctional ALAS2 enzyme presumably do not develop into viable erythrocytes, so that the patients’ circulating red cells represent progeny of the residual normal clone released from marrow at an accelerated rate in response to the anemia. Some females with milder anemia have a biphasic red cell volume histogram characterized by populations of normocytic and microcytic cells. Nonanemic female carriers of this trait may or may not have a small microcytic circulating erythrocyte population, but when present, may suggest the correct diagnosis in a related male proband. The morphologic features of the anemia in individuals affected by SLC25A38 mutations are indistinguishable from males with XLSA. Nonetheless, it is exceptional for XLSA to present in early childhood, and, consequently, mutations in SLC25A38 should be excluded in all individuals presenting with microcytic CSA in infancy or early childhood. As is true of all CSAs, the definitive diagnosis rests on showing a genetic defect by mutational analysis (see Table 1 ).
Syndromic CSAs
The presence of nonhematologic manifestations distinguishes the syndromic CSAs from the nonsyndromic CSAs. Sometimes, the associated features may be subtle or not yet fully manifest at the time of presentation, making recognition on clinical grounds elusive.
XLSA with ataxia (XLSA/A), described in 4 kindreds to date, is characterized by mild to moderate microcytic SA accompanied by neurologic deficits of delayed motor and cognitive development, incoordination, and cerebellar hypoplasia. Female carriers of XLSA/A show mild hematologic abnormalities, including an increased RDW and occasional peripheral blood siderocytes, but no neurologic manifestations. XLSA/A is caused by mutations in the mitochondrial adenosine triphosphate–binding cassette transporter ABCB7, which is postulated to participate in the export of Fe-S clusters generated in mitochondria for assembly of cytosolic Fe-S cluster-containing proteins. Like mutations in GLRX5, it is presumed that ABCB7 dysfunction disturbs cellular and mitochondrial iron metabolism through its effects on IRP1, among other proteins.
The syndrome of CSA, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD) presents in infancy and is also variably associated with central nervous system (CNS) abnormalities, sensorineural hearing loss, and cardiomyopathy. The anemia is microcytic and severe, often requiring chronic transfusion or stem cell transplantation. Inheritance is autosomal recessive.
The Pearson marrow-pancreas syndrome generally presents within the first 6 months of life with failure to thrive, anemia ± other cytopenias, metabolic acidosis, and exocrine pancreatic insufficiency; hepatic and renal failure are also common. Absence of the metabolic derangements initially may result in oversight of the syndrome, particularly when ring sideroblasts are rare or when there is a marked erythroid hypoplasia, in which case the disorder may be mistaken for Diamond-Blackfan anemia. The anemia tends to be severe and is most often macrocytic. In the bone marrow, there is striking vacuolization of erythroid and myeloid progenitors. In skeletal muscle there are marked abnormalities in mitochondrial ultrastructure, most characteristically parking lot inclusions in mitochondria, which represent linear arrays of cristae. Deletions (most often of 4977 bp, which is common to many mitochondrial cytopathies), rearrangements, or duplications of mitochondrial DNA (mtDNA) are diagnostic. The heteroplasmy of mtDNA at the cellular and organ levels accounts for the high variability of affected tissues, which may even evolve over time. There is no single common deleted region in all patients. Instead, it seems that 1 or several mitochondrially encoded transfer RNAs (tRNA) are deleted in nearly all cases, and would be expected to lead to a global defect in mitochondrial protein translation. As in several other forms of CSA, the mechanism for the mitochondrial iron accumulation is not understood.
The central role of mitochondrial tRNAs and mitochondrial translation in the pathogenesis of several of the syndromic CSAs is reinforced by mitochondrial myopathy, lactic acidosis, and SA (MLASA), which is an autosomal recessive disorder encountered in infancy, childhood, or adolescence. The cardinal clinical findings are skeletal muscle weakness, variably severe normocytic anemia, and lactic acidosis. Vacuolization of marrow progenitors and ultrastructural findings on skeletal muscle electron microscopy are indistinguishable from Pearson marrow-pancreas syndrome. Mutations in 2 genes result in the MLASA phenotype. Pseudouridine synthase 1 (PUS1) posttranscriptionally modifies cytosolic as well as mitochondrial tRNAs by converting uridine to pseudouridine, stabilizing the tRNA secondary structure. Mitochondrial tyrosyl tRNA synthetase (YARS2) charges the mitochondrially encoded tyrosine tRNA with its cognate amino acid. Missense and nonsense mutations in either protein presumptively lead to decreased mitochondrial protein synthesis and respiratory chain dysfunction, but how these mitochondrial protein deficiencies cause mitochondrial iron accumulation and anemia is unclear.
Thiamine-responsive megaloblastic anemia (TRMA) syndrome typically manifests as a triad of megaloblastic anemia, non–type I diabetes mellitus, and sensorineural deafness between infancy and adolescence. The macrocytic anemia is associated with variable neutropenia and thrombocytopenia and megaloblastic changes. The molecular diagnosis is established by biallelic mutations in the SLC19A2 gene, which encodes a high-affinity thiamine transporter. Most mutations are nonsense or frameshift mutations leading to complete loss-of-function alleles, but increasingly, missense alleles have been identified in individuals with less overt TRMA phenotypes. The macrocytic megaloblastic anemia is considered to result from defective nucleic acid synthesis attributed to cellular thiamine deficiency. The ring sideroblasts are hypothesized to relate to the role of thiamine in the production of succinyl-coenzyme A, a substrate of ALAS.
Undefined CSAs
Up to 40% of CSA cases are molecularly unexplained. In these cases, autosomal recessive as well as X-linked defects are possible and may be shown in previously identified genes causing the phenotype with other approaches of mutational analysis. Alternatively, within the presently understood pathophysiologic framework of iron homeostasis in the erythroid cell, novel genes involving heme or Fe-S cluster production, as well as mitochondrial proteins that affect iron processing or utilization, may be discovered.
Acquired Clonal SAs
This most common SA encountered in clinical practice develops insidiously in middle-aged and older individuals. It may be discovered during a routine examination or in association with an unrelated complaint. The anemia is usually of moderate severity, normocytic or macrocytic, but dimorphic because of the presence of a hypochromic erythrocyte population on the blood smear.
The disorder is a bone marrow stem cell disorder and classified within the rubric of myelodysplastic syndromes (MDS) and myeloproliferative neoplasms. In the current World Health Organization classification of hematopoietic neoplasms, 3 variants are distinguished: refractory anemia with ring sideroblasts (RARS), RARS with thrombocytosis (RARS-T), and refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS). Classification into each of these forms, as the names indicate, depends on the presence or absence of dysplasia within the nonerythroid marrow lineages as well as the clinical absence or presence of thrombocytosis. As is described later, there is increasing molecular evidence that these disorders are on a continuous phenotypic spectrum that is related to primary driver somatic mutations in a restricted class of genes, on which secondary mutations supervene to modify the morphologic and clinical phenotype.
In early work focusing on heme biosynthesis no consistent defects in protoporphyrin synthesis in marrow cells were found. A somatic ALAS2 mutation was identified in only 1 case. However, a constant feature is mild to moderate increase of erythrocyte protoporphyrin, and impaired marrow FECH activity was observed in about one-half of patients studied. Uncommonly, clinical or biochemical features resemble EPP, which may be explained by a likely acquired deletion of an FECH allele caused by a cytogenetic abnormality of the clonal disorder.
Subsequently, intraclonal heterogeneity of the erythroblast phenotype (ie, a variable amount of mitochondrial iron overload evident from cell to cell) had suggested a heteroplasmic state caused by a mitochondrial defect(s). Extensive analysis of mtDNA in patients with RARS as well as other MDS forms showed a wide spectrum of heteroplasmic mutations across the mitochondrial genome in approximately one-half of cases. Although their functional importance overall remains unclear, some mutations may affect mitochondrial iron metabolism (eg, if they affect cytochrome oxidase).
In recent years, mutations in protein constituents of the spliceosome, which mediates maturation of primary mRNA transcripts into mature mRNAs lacking introns, have been recognized as being common in MDS. Specifically, acquired heterozygous missense alleles of the SF3B1 (splicing factor 3B, subunit 1) component of the splicing machinery are present in up to 85% of patients with RARS, RARS-T, and RCMD-RS. The presence of multiple specific recurrent alleles localized to a restricted portion of the SF3B1 protein suggests that these mutations are not loss-of-function alleles, but rather gain-of-function variants or mutations that affect the function of the protein in an otherwise distinctive manner. In cases lacking an SF3B1 mutation, other spliceosome components, particularly SRSF2 or ZRSR2, are commonly mutated. Among the myriad of genes whose splicing and expression may be altered by mutations in the spliceosome, there is evidence to suggest that changes in ABCB7 expression may be the downstream mechanism(s) leading to the dysregulated iron metabolism and sideroblastic phenotype.
The diagnosis of MDS with ring sideroblasts must be established by exclusion of other causes of SA. These conditions are rare, if they occur at all, in children and adults less than 40 years of age, in whom a congenital basis, and less likely a metabolic cause (see later discussion), should be considered. Moreover, because the hematologic phenotype of females expressing XLSA is indistinguishable from RARS, analysis of the ALAS2 gene for mutations should be entertained in them.
Acquired Metabolic SAs
Exposure to ethanol and certain drugs, as well as acquired copper deficiency, may lead to an SA with features that overlap with those of the congenital or acquired clonal forms. In these instances, the anemia is fully reversible when the offending factor is removed.
Ethanol, by disrupting heme synthesis, suppressing erythroid colony formation, and likely inhibiting mitochondrial protein synthesis, contributes to the usual multifactorial anemia associated with alcoholism. The ring sideroblast abnormality is noted in up to one-third of anemic alcoholic patients and most characteristically in the presence of malnutrition and folate deficiency. The MCV is normal or increased; dimorphic erythrocytes are common, and siderocytes may be present on the blood smear. A highly suggestive finding in bone marrow is vacuolization of pronormoblasts.
Although isoniazid and chloramphenicol have been the prototypical drugs that produce an SA, a series of other agents have been implicated and include cycloserine, pyrazinamide, linezolid, fusidic acid, busulfan, melphalan, penicillamine, and triethylenetetramine dihydrochloride. Many antituberculosis drugs interfere with vitamin B 6 metabolism and impair heme synthesis. Linezolid, a more recently used antibiotic, induces the ring sideroblast abnormality, and its toxicity resembles that of chloramphenicol because it also inhibits mitochondrial protein synthesis although by a different mechanism. The association with the other drugs was reported in single or in a few patients, and their toxicity in the erythron is not characterized.
Nutritional copper deficiency occurs when the metal is omitted in parenteral feedings, in malabsorption states, and after gastrointestinal resections. Another cause is prolonged ingestion of zinc (eg, in zinc supplements, zinc-containing denture cream, coins). Zinc induces metallothionein, resulting in sequestration of copper in the intestinal epithelium, and decreased absorption of copper in the gut. Various neurologic manifestations, including CNS demyelination, peripheral neuropathy, optic neuropathy, and most often myeloneuropathy, are usually, if not invariably, present and, in time, become irreversible. The anemia may be severe, the MCV is normal or increased, and neutropenia is frequent. Low serum copper and ceruloplasmin levels confirm the diagnosis.
Disease courses and treatment options
The clinical courses of these heterogeneous disorders are highly dependent on the underlying cause as well as on the supportive care that the patient receives, particularly if the disease is severe.
Nonsyndromic CSA
In XLS, the anemia is usually mild to moderate in severity and remains so at the set point determined by the mutation-induced functional impairment of the ALAS2 enzyme. In up to two-thirds of cases, the anemia responds to the essential cofactor of ALAS in the form of pyridoxine supplements by enhancing the function of some ALAS2 mutants. On average, the hemoglobin level normalizes in about one-third of responders. Uncommonly, severe anemia that is unresponsive to pyridoxine dictates transfusion dependence, with attendant accentuation of iron overload. This finding has also been observed in females expressing XLSA caused by severe ALAS2 mutations and progressive inactivation of the normal X chromosome with advancing age.
Patients with the uniformly severe anemia caused by SLC25A38 defects require regular transfusions lifelong. Hematopoietic stem cell transplantation has been successful in 5 unreported cases. In the single patient with GLRX5 deficiency, transfusions were believed to worsen the anemia, which was partially reversed by iron chelation with deferoxamine. The mild anemia associated with EPP requires no treatment, but the patients’ lifelong course of variable photosensitivity may be abruptly complicated by progressive liver disease in ∼2% of cases, requiring liver and/or bone marrow transplantation.
Syndromic CSA
The clinical course in the XLSA/A syndrome is dominated by the nonhematologic features of neurologic changes and is dependent on supportive or rehabilitative services.
The severity and progressive nature of SIFD has led to early death from cardiac or multiorgan failure in 53% of cases. Supportive treatment has included regular transfusions and immunoglobulin infusions in most patients. Hematopoietic stem cell transplantation was successful in one, correcting the anemia and immunodeficiency.
Similarly, about one-half of patients with Pearson marrow-pancreas syndrome succumb to the associated metabolic derangements. The anemia improves in survivors or they develop Kearns-Sayre syndrome.
Patients with the MLASA syndromes have had unpredictable clinical courses. Manifestations at presentation have been highly variable, even within or between families with the same genetic variant, and some patients with YARS2 defects have shown spontaneous improvement. Severe disease, also manifesting cardiomyopathy, portended demise in 2 cases at ages 3 months and 18 years. Many patients have survived into adulthood.
In the TRMA syndrome phenotypic variability from the outset influences the prognosis. Thiamine (vitamin B 1 ) is prescribed in pharmacologic doses and usually improves the anemia and the diabetes, although it has become ineffective in adulthood.
Acquired Clonal SA
The natural history of the RARS and RARS-T variants is typically a stable, nonprogressive anemia of many years duration. Transfusions may be necessary for symptomatic anemia, in particular at advanced age or in the presence of other comorbid conditions. Patients with RCMD-RS tend to have more severe anemia, have decreased survival, and ∼5% evolve into acute leukemia.
Various agents used in therapeutic trials for MDS, which have included the ring sideroblast variants, have been erythropoietin with or without granulocyte colony-stimulating factor and various drugs (eg, 5-azacytidine and decitabine, etanercept, antithymocyte globulin, thalidomide and lenalidomide, and valproic acid). On average, major responses with improved erythropoiesis have been limited or less than 50%.
Splenectomy in SA
This operation has often been considered and occasionally performed in patients with CSA with severe anemia and significant splenomegaly. Because this procedure is invariably complicated by postoperative thromboembolic disease and often a fatal outcome, it should be considered contraindicated. Factors other than persistent thrombocytosis seem to play a role, and control of the platelet count and anticoagulant therapy are usually ineffective.
Iron overload
Systemic iron overload, also termed erythropoietic hemochromatosis, develops to a variable degree in several forms of SA (see Table 1 ), but most prominently in CSA, and it can significantly contribute to morbidity and mortality. Mild to moderate hepatosplenomegaly with preserved liver function is common. The iron deposition in liver is indistinguishable from hereditary hemochromatosis, being predominantly hepatocellular and periportal in nature. In XLSA, the iron burden has not always correlated with the severity of the anemia and well-established cirrhosis may be discovered in the third or fourth decade even in patients not known to be anemic. Hepatocellular carcinoma has developed in 2 reported cases. In time, cardiac arrhythmias and congestive heart failure supervene. In children, growth and development may be impaired. The process is not HFE linked, although occasionally it may be accentuated by coinheritance of an HFE-hemochromatosis allele(s).
This form of iron overload is driven by the ineffective erythropoiesis so typical of SA (as well as thalassemia and congenital dyserythropoietic anemia) by mediating inappropriately increased intestinal absorption of iron through suppression of the iron regulatory hormone hepcidin by an ill-defined mechanism. Mediators such as GDF-15 (growth differentiating factor 15) and TWSG1 (twisted gastrulation) released from the ineffective erythron in the bone marrow have been postulated. More recently, Erfe (erythroferrone) has been reported as the erythroid factor that represses hepcidin during increased erythropoietic activity. However, the long sought after erythroid regulator of iron metabolism as first proposed by Finch has not yet been definitively ascertained.
Repeated transfusions for severe anemia predictably add to the iron burden, because each unit of red cells delivers 200 to 300 mg of iron. Although transfusional iron overload may at least in part result in nonparenchymal distribution of the iron, it can significantly add to morbidity and affect survival. As extensively shown in thalassemia, long ago, and, more recently, in limited studies in MDS, mortality can be improved with effective iron-chelation therapy. Chelation limits nontransferrin-bound iron and labile iron pools in plasma and cells rapidly, whereas removal of iron stored in tissues is mobilized more slowly. However, considerable debate persists regarding precise indications for chelation therapy in transfusional iron overload, which also seemed to be reflected at least partly in the management variation of a large patient group. Although the MDS Foundation’s Working Group on Transfusional Iron Overload and other groups have developed consensus statements on the issue, data from prospective trials are desired. Meanwhile, the decision regarding iron-chelation therapy in this setting is best based on current consensus statements as well as on a case-by-case basis.
Iron depletion is best performed by phlebotomy when the anemia is mild or moderate (ie, hemoglobin ≥9 g/dL), or when a patient with XLSA has responded to pyridoxine. After initial iron depletion, regular phlebotomies are continued for life to control iron reaccumulation. In patients with more severe anemia and who require regular red cell transfusions, iron chelation is used. The available agents in the United States for treatment of all transfusional iron overload disorders, including SA, are deferoxamine (Desferal, Novartis) for parenteral administration and deferasirox (Exjade, Novartis) as the oral preparation. A third chelating agent, deferiprone (Ferriprox, Apotex) is licensed for use in thalassemia with a second-line indication. Combinations of these drugs can be used in severe iron overload, although trials of such therapy in SA have not been performed and would be considered off label. Anemia may improve after adequate iron removal. An independent salutary effect of iron chelation on erythropoiesis was also implicated in some cases.
Summary
Although SA is uncommon and some forms are rare, it should be considered in all adults and children/infants with unexplained anemia of any severity from history, clinical examination, and basic laboratory data. Certain clinical features are suggestive, such as history of chronic anemia; exposure to factors causing reversible SA; family history of anemia; presence of neurologic abnormalities; myopathy, lactic acidosis, immunodeficiency in children or young adults.
If the pathognomonic ring sideroblasts are evident on a Prussian blue stain of the bone marrow aspirate smear, a careful review of the patient’s constellation of clinical findings and the erythrocyte indices and morphology aid in narrowing the differential diagnosis ( Fig. 3 , see Table 1 ). Erythrocyte morphology is most accurate before any transfusion, which can conceal the abnormalities (eg, a microcytosis and hypochromia). When the causative gene is known, mutation analysis provides the definitive diagnosis of a CSA. Some molecular genetic tests are available in several clinical laboratories, whereas others are available only in certain research laboratories. In cases of CSA in which the genetic cause is identified, a search for affected family members is advisable. In the common forms of SA, evaluation and monitoring for iron overload are indicated by biochemical testing (serum transferrin saturation and ferritin) and noninvasive assessment of parenchymal organ involvement by magnetic resonance imaging.