Chapter Outline
Effects on the Erythrocyte Membrane
External Membrane Interactions
Hemolysis and Nitric Oxide Depletion
Acute and Chronic Inflammation
Sickle Cell Trait (α 2 β 2 , α 2 β 2 6 Val )
Hemoglobin SC Disease (α 2 β 2 6 Val , α 2 β 2 6 Lys )
Hemoglobin SO Arab (α 2 β 2 6 Val , α 2 β 2 121 Lys )
Hemoglobin SD (α 2 β 2 6 Val , α 2 β 2 121 Glu )
Hemoglobin S Korle Bu (α 2 β 2 6 Val , α 2 β 2 73 Asn )
Hemoglobin C Harlem (C Georgetown ) (α 2 β 2 6 Val,73 Asn )
Hemoglobin S Antilles (α 2 β 2 6 Val,23 Ile )
Hemoglobin S Québec-CHORI (α 2 β 2 6 Val , α 2 β 2 87 Ile )
Hemoglobin SE (α 2 β 2 6 Val , α 2 β 2 26 Lys )
The first report of sickle cell anemia (SCA) in the medical literature described a dental student from Grenada, Walter Clement Noel. It was by Herrick and Irons in 1910 and introduced the word sickle in the title of the case report. Sydenstricked described the first cases in children, recognized the association with hemolytic anemia, and introduced the term crisis to describe periodic acute episodes of pain. The pathologic basis as a disorder of hemoglobin (Hb) was defined in 1927, confirmed 2 decades later by electrophoresis that separated sickle Hb from normal Hb. Neel defined the genetics of the disorder and clearly distinguished sickle cell trait—the heterozygous condition (AS)—from SCA, the homozygous state (SS).
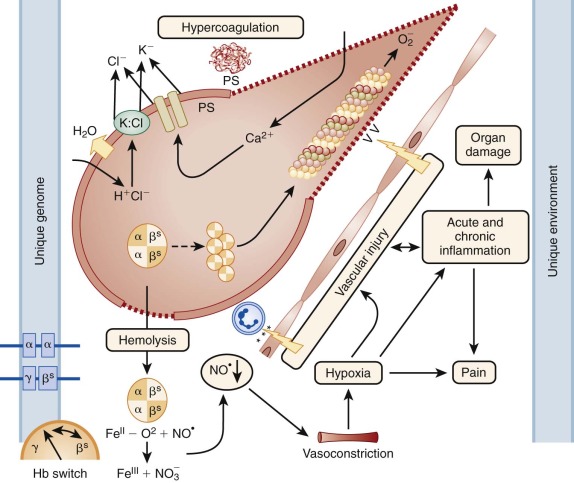
Further understanding of the molecular basis of the disorder was made possible by the finding that normal human Hb is composed of two pairs of globin subunits. Beyond the embryonic period, one globin pair is invariant (α chain), and the other pair is variable (γ, δ, or β chain). The amino acid substitution in sickle cell Hb is now known to be valine for glutamic acid in the number 6 position of the β chain (β S ). Figure 20-1 provides an overview of pathophysiology of β S mutation).
In this chapter, sickle cell disease (SCD) refers to the broad group of disorders that feature erythrocyte sickling under physiologic conditions. SCD genotypes include homozygous HbSS and compound heterozygous conditions including HbS β 0 -thalassemia, HbS β + -thalassemia, and HbSC disease. HbSS, the most prevalent genotype, and HbS β 0 -thalassemia are clinically similar and therefore are commonly referred to as SCA ; these genotypes are associated with the most severe clinical manifestations. Persons with sickle cell trait (HbAS), the carrier state, rarely have clinical problems and are not considered to have a form of SCD.
Pathophysiology
Hemoglobin S Mutation
The sickle gene mutation is common in sub-Saharan Africa and other parts of the world with endemic malaria. The allele frequency of the β S mutation closely tracks the distribution of malaria worldwide. Persistence and expansion of such a presumably deleterious mutation is explained by the benefits conferred in the heterozygous state; persons with SCT have a survival advantage from malaria.
Polymorphisms that flank the β S mutation indicate that this mutation developed independently and spontaneously at least five times. Within sub-Saharan Africa, there are four major sickle haplotypes, each associated with a particular geographic region: Senegal, Benin, Central African Republic (CAR), and Cameroon. Among North American patients of African heritage with sickle cell, 50% to 70% of chromosomes are Benin, 15% to 30% are Bantu-CAR, and 5% to 15% are Senegal. Benin and Senegal haplotypes are associated with higher levels of fetal Hb (HbF) and fewer dense cells compared with CAR, but most African Americans are compound heterozygotes. As a result, these haplotypes have only limited predictive value for overall clinical severity, with CAR being the most severe and the Senegal variety being the least severe.
A different haplotype is found in India and eastern parts of Saudi Arabia and is known as the Arab-Indian haplotype. Patients from these regions have long been recognized as having milder disease, particularly in childhood, with elevated levels of HbF. However these patients develop chronic organ damage that may reflect, in part, their higher Hb concentrations.
Heterozygotes with HbS, HbC, HbE, α- and β-thalassemia and glucose-6-phosphate deficiency (G6PD), particularly infants, are partially protected from cerebral falciparum malaria relative to the normal population. The mechanistic basis for this survival advantage from the sickle gene is not fully understood, but as recently reviewed, the effect is multifactorial with impaired parasite growth and development, enhanced clearance of infected cells from the circulation, and altered adherence of damaged erythrocytes to the capillary endothelium.
The Hemoglobin S Polymer
Polymer Structure
In 1927, Hahn and Gillespie showed that red blood cell (RBC) sickling, the change from a biconcave disk to the sickle form, was dependent on deoxygenation. Harris subsequently demonstrated that cellular sickling was associated with the formation of “tactoids” of HbS that appeared as the Hb became deoxygenated. Electron micrographs of sickled red cells reveal long, thin bundles of HbS fibers that run parallel to the long axis of the cell. The ultrastructure of HbS fibers, as detailed by electron microscopy and image reconstruction, reveal a complex solid-core structure 21 nm in diameter composed of 14 filaments arranged as seven pairs of double filaments.
The detailed crystal structure suggested by Wishner and Love and colleagues identified several critical intermolecular contact sites. Contacts along the axis of the filament are made by α and β chains, whereas lateral contacts between filaments of a pair are largely between β chains and contacts between filament pairs are largely through α chains. Within each Hb tetramer one abnormal β chain contributes the β S mutation, whereas the other contributes a critical receptor region around the Phe β85 residue. In this critical receptor region lie residues where mutants affect fiber formation: β73 (Korle Bu), β66 (I Toulouse), β83 (Pyrgos), and β87 (D Ibadan). Within the acceptor pocket the mutant valine closely contacts four different hydrophobic residues (β70, β73, β84, and β85). In addition to the hydrophobic interaction, water molecules that form hydrophilic interactions in the lateral contact region are present near the mutant valine.
Polymer Formation
The polymerization of deoxygenated HbS (deoxy-HbS) is a highly complex process that results in the formation of gelled, aggregated HbS tetramers in equilibrium with Hb tetramers in solution. Perturbations in oxygen levels, temperature, pH, ionic strength, 2,3-diphosphoglycerate (2,3-DPG), and carbon monoxide affect the formation of HbS gels. This intracellular polymerization of HbS is the sine qua non of the disorder and leads to distorted cell morphology, altered blood viscosity, circulatory sludging with occlusion of blood flow, and ultimately tissue damage and organ infarction that cause clinical manifestations of SCD.
The kinetics of HbS polymerization can be explained by a double nucleation mechanism. Gelation is initiated by a process called homogeneous nucleation, in which single deoxy-HbS molecules aggregate. Aggregation of a few molecules is thermodynamically unstable, but once a certain number of molecules aggregate, a condition termed the critical nucleus, addition of further molecules produces a more stable aggregate or polymer. Thus homogeneous nucleation is highly dependent on the concentration of deoxy-HbS molecules. Gelation continues with heterogeneous nucleation that occurs on the surface of preexisting polymer. The same intermolecular contacts between the mutant valine and its receptor on the surface of the polymer are responsible for heterogeneous nucleation and cross-linking between strands. While polymerization progresses, more surface area becomes available and therefore the reaction becomes autocatalytic.
The result of this double nucleation mechanism is a measurable delay time between the initiation of polymerization and the exponential rise in polymer formation. In solution, the delay time varies as the thirtieth power of the Hb concentration: 1/t d = K (C/C s ) n where t d = delay time, C = Hb concentration, C s = Hb solubility, and n = 30 (the number of Hb tetramers in the critical polymer). Because n is so large, very small changes in Hb concentration have a profound effect on the delay time. This phenomenon of delayed gelling of HbS in solution is also observed in erythrocytes containing HbS. The distribution of observed delay times within intact RBCs is consistent with the distribution of the mean corpuscular Hb concentration (MCHC). Slow deoxygenation causes large aligned polymers, which results in significant distortion of cell morphology.
A kinetic model of HbS gelation that incorporates the concept of a critical delay time led Eaton and co-workers to propose that polymerization kinetics play an important role in the pathophysiology of SCA ( Fig. 20-2 ). Erythrocytes exposed to oxygen within the lungs are quickly “degelled” so the arterial circulation contains little polymerized HbS and few sickled cells. But as the cells enter the capillary circulation, the oxygen saturation (SpO 2 ) decreases rapidly and the HbS solubility also drops. If the transit time within the capillary (analogous to the delay time) is longer than 1 second, the cell will sickle and likely occlude the capillary, whereas a fast transit time means sickling will not take place in the capillary and obstruction will not occur. Patients who become hypoxic, acidotic, dehydrated, and febrile are likely to experience vaso-occlusive episodes caused in part by a shorter delay time. The hypertonic renal medulla and the acidotic, high-hematocrit environment of the spleen make these organs prime targets for sickling.

Relatively little is known about the uniformity of the depolymerization process within erythrocytes, but failure to fully melt the polymer within the lungs may lead to residual polymer. Important variables that affect HbS polymer formation (HbF, 2,3-DPG levels, SpO 2 , and pH) are heterogeneously distributed among erythrocytes, and these factors (along with membrane abnormalities and red endothelial interactions) likely play a role in the variable clinical severity of SCD.
Interactions of Hemoglobin S with Hemoglobin A and Hemoglobin F
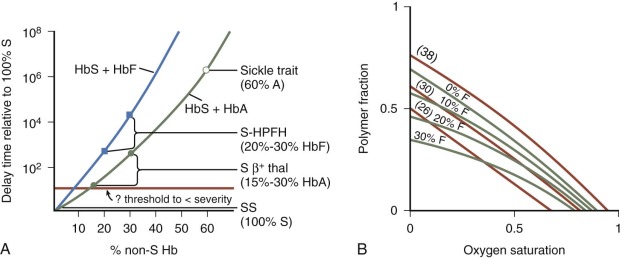
The study of the interaction between HbS and other Hbs supports a rational basis for understanding the clinical manifestations of various sickle syndromes and provides a rationale for therapy. Investigators have extensively studied mixtures of HbS with HbF or HbA to determine the effects on gelation and solubility. The kinetic data show that both HbA and HbF have a profound, dose-related effect, increasing the delay time and decreasing HbS polymer content within erythrocytes ( Fig. 20-3 ), but the effects of HbF are greater than HbA. Compared with pure HbS solutions, mixtures with 15% to 30% HbA (as found in HbS β + -thalassemia) have delay times that are up to 100 times longer; mixtures with 20% to 30% HbF (as found in HbS–hereditary persistence of fetal hemoglobin [HPFH]) have delay times that are up to 10 4 times longer, and mixtures with 60% HbA (as found in sickle trait) have delay times that are 10 6 times longer. HbA and HbF also increase the solubility of HbS, with HbF again being more effective than HbA. Very little if any HbF is incorporated into the polymer, suggesting that asymmetric hybrids of HbS and HbF (α 2 β S γ) are not incorporated into polymer under most physiologic conditions, whereas asymmetric hybrids of HbS and HbA (α 2 β S β A ) are readily incorporated into the HbS polymer.
Effects on the Erythrocyte Membrane
The basic pathophysiology of SCA is directly related to HbS polymerization. However effects of polymerization on the erythrocyte membrane, which is in direct contact with this intracellular process, alter the pathophysiology of the disease. For example when the damaged membrane breaks, then intravascular hemolysis will ensue. When the altered membrane interacts with other circulating blood cells, soluble plasma factors, the vascular endothelium, and the reticuloendothelial system (RES), a wide range of additional events can lead to the highly variable but often severe phenotypic expression.
Mechanisms of Membrane Damage
In normal erythrocytes, HbA binds to the membrane around band 3 protein and also to phospholipids on the inner surface of the membrane. Hb at physiologic concentrations also stabilizes the configuration of spectrin heterodimers. HbS binds to membranes more readily than HbA and membrane-associated sickle Hb is a major determinant of erythrocyte rigidity. Membrane rigidity has been associated with a small amount of high–molecular-weight spectrin-Hb complex in the membrane.
HbS readily denatures and forms small aggregates, so-called micro-Heinz bodies that attach with high affinity to the cytoplasmic portion of band 3 protein at the HbA binding site. Both band 3 protein and glycophorin are clustered above these micro-Heinz bodies, and clustering of band 3 protein is associated with the deposition of specific IgG antibodies onto the cell, which are linked to normal cell senescence and may contribute to shorten the circulating half-life of HbS erythrocytes. Ankyrin is also abnormally clustered around the denatured HbS, and like band 3 protein, may be damaged in the process.
Membrane damage also occurs when HbS auto-oxidizes to form methemoglobin (met-Hb), thereby generating superoxide and losing heme. Intracellular iron from different compartments (e.g., denatured Hb, free heme, hemichromes, and nonheme iron) can bind to the inner membrane and serves as a catalyst for production of highly reactive hydroxyl radicals that cause oxidative damage to the membrane. Erythrocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity was recently identified as a source of reactive oxygen species (ROS) that contribute to erythrocyte rigidity and fragility in SCA. Therapies directed toward ROS may therefore be beneficial in SCA by reducing oxidative stress.
Membrane Deformability
While sickle erythrocytes become deoxygenated and fill with polymerized HbS they become less deformable. The viscoelastic properties of sickled erythrocytes can be measured by ektacytometry, which allows investigation of the deformability of HbSS erythrocytes in aggregate and has shown that decreased deformability is directly related to the amount of intracellular Hb concentration. Using micropipette techniques to measure membrane deformability of individual cells, two studies have shown that the mean corpuscular hemoglobin concentration (MCHC) is the major contributor to cell rigidity, although cellular dehydration independently increases membrane rigidity.
Cell rigidity with lack of deformability is further evidence that a structural membrane lesion is present. Most erythrocytes undergo repeated HbS polymerization/depolymerization that leads to repeated shape change from the characteristic sickled form to the normal biconcave disc, and thus represent reversibly sickled cells (RSCs). However some erythrocytes retain the sickled conformation even upon reoxygenation; the cytoskeleton of such irreversibly sickled cells (ISCs) has a variety of abnormalities including aberrant cross linking of actin, dissociation of spectrin tetramers, and clustering of band 3 protein and glycophorin. Disordered mobility of the cytoskeleton relative to the overlying membrane may affect protein–protein interactions including binding of normal spectrin to sickle ankyrin. With proteomic techniques four groups of proteins are increased in sickle erythrocyte membranes: actin accessory proteins, components of lipid rafts, scavengers of oxygen radicals, and protein repair participants. All of these observations support the hypothesis that membrane rigidity is the result of cytoskeletal damage associated with HbS polymer formation and oxidative damage.
Hemoglobin Concentration
Patients with SCA have a wide variation in their erythrocyte density, including many light cells (reticulocytes) and more dense cells (MCHC >37 g/dL) than normal erythrocytes. This is reflected in the histograms of mean corpuscular volume (MCV) and Hb from the sickle genotypes ( Fig. 20-4 ). Sickle erythrocytes separated by density (and therefore MCHC) obstruct flow in an artificially perfused capillary system in proportion to their intracellular Hb concentration. However there appears to be no correlation between the percentage of dense cells and the frequency or onset of vaso-occlusive events. A newly described in vitro system with microfluidics and endothelialized channels may represent an improved method to study the rheological characteristics of sickled erythrocytes and will aid the investigation of pathologic consequences of ISC and dense cells as they interact with vascular endothelium.
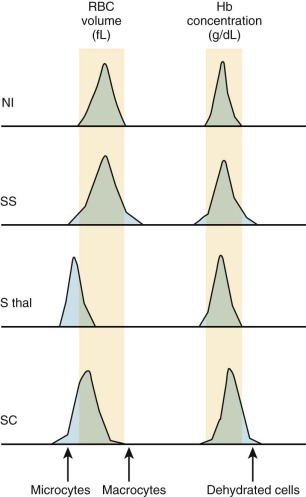
The heterogeneity of red-cell shape and density in SCA is influenced significantly by the distribution of HbF within the erythrocytes. For unexplained reasons, HbF is not evenly distributed among erythrocytes (both in normal persons and patients with SCA) and defines a subpopulation known as F cells. Because HbF helps prevent HbS polymerization, F cells sickle less often and thus survive longer in the circulation. The densest cell fraction in SCA contains the oldest erythrocytes that contain the lowest amounts of HbF and include the ISC fraction.
Erythrocyte Shape Changes
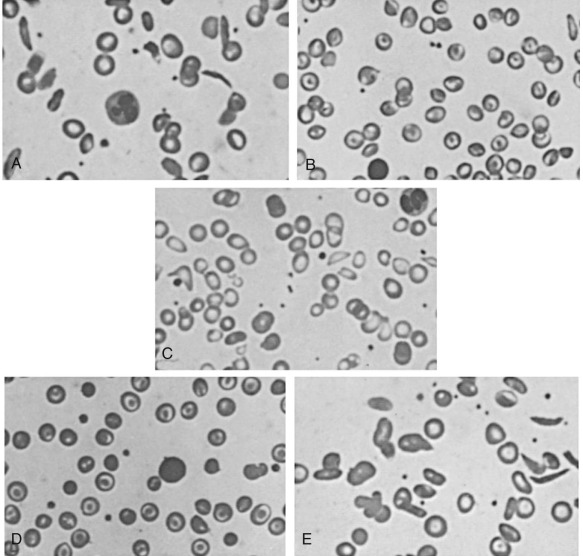
RSCs have normal shape and normal viscosity when oxygenated. The vaso-occlusive complications of SCD may be because of the fact that these cells are able to circulate into the microvasculature because of their normal rheologic properties when oxygenated but then become distorted and more viscous as they become deoxygenated within the small capillaries. In contrast ISCs are slender, elongated cells with blunt ends that are visible even on an oxygenated peripheral blood smear ( Fig. 20-5 ). These cells are extremely dense with MCHC of greater than 44 g/dL, have little HbF to dilute their concentrated HbS, and survive in the circulation for only a few days. ISCs can be formed in vitro by prolonged deoxygenation and by repeated oxygenation–deoxygenation cycles. Reversible oxidative damage to β-actin and loss of ubiquitination of α-spectrin leads to formation of an ISC membrane skeleton that is “locked” because it cannot disassemble or reassemble. Antioxidants can reduce the formation of ISCs in vitro. ISC numbers vary widely from patient to patient and have been correlated with hemolysis and splenomegaly but only poorly with vaso-occlusive severity.
Ion Transport and Cell Volume
Abnormal cation permeability of the sickle erythrocyte is characterized by calcium loading, potassium depletion, acidosis, and dehydration, all of which enhances the tendency for HbS polymerization (see Fig. 20-1 ). HbS polymer formation upon deoxygenation causes a reversible potassium loss and sodium gain in the cell. Abnormal water and cation movements across the cell membrane have been associated with a variety of pumps, channels, and leaks involving potassium (K + ), sodium (Na+), chloride (Cl − ), calcium (Ca 2+ ), and magnesium (Mg 2+ ). Although the pathogenesis of these pathways remains incompletely understood, they are undoubtedly related to the physical distortion associated with sickling, oxidant damage, and the Hb mutation itself.
At least two major erythrocyte membrane channels are responsible for cellular dehydration: the calcium-activated potassium-selective process (Gardos channel, encoded by the KCNN4 gene) and the K + Cl − cotransporter (encoded by KCC with multiple isoforms). These channels are critical for cation flux and water regulation in the sickle erythrocytes, and thus influence Hb concentration and cell volume. Sickle reticulocytes exposed to deoxygenation and HbS polymerization accumulate sufficient Ca 2+ to activate the Gardos channel, which results in osmotic-driven water loss and rapid RBC dehydration to a much greater extent than with other transporters. The Gardos channel can be inhibited by the antimycotic agent clotrimazole, and a short-term study demonstrated benefits on cellular hydration. A novel Gardos channel inhibitor (senicapoc, ICA-17043) entered into formal clinical trials, and although having substantial effects on cellular hydration, a definitive phase III double-blinded placebo-controlled trial demonstrated no efficacy for reducing vaso-occlusive events.
A second pathway involved in RBC dehydration is the potassium chloride channel (KCC). The KCC is active in reticulocytes as part of normal volume reduction during cellular maturation but has increased activity in sickle reticulocytes with deleterious consequences. The KCC pathway is stimulated to lose K + , Cl − , and water when predominantly young sickle erythrocytes are exposed to a low pH environment, as occurs in areas of poor perfusion. The KCC pathway is also activated by deoxygentation of Hb and by low levels of Mg 2+ . Genes for four KCC isoforms have been identified; KCC1 , 3 , and 4 isoforms are present in normal and sickle erythrocytes, but the latter may have N-terminal splicing variants of these isoforms. The KCC pathway can be inhibited by magnesium in vivo, and Mg 2+ is depleted in sickle erythrocytes possibly through an abnormal Na–Mg exchange. Preliminary studies demonstrated the effectiveness of oral magnesium in the prevention of cellular dehydration, and a pilot trial suggested utility for intravenous (IV) magnesium in the setting of acute vaso-occlusive pain.
External Membrane Interactions
Increased Erythrocyte Adhesion to the Endothelium
In 1979 investigators first discovered that sickle erythrocytes have an abnormal propensity to adhere to vascular endothelial cells with an adhesive force that results from numerous attachment sites that withstand detaching forces found in low shear vascular beds. While cells flow through vessels of critical dimension, abnormal flow caused by endothelial interactions increases the odds that polymerization and obstruction will occur (see Fig. 20-1 ). Support for this hypothesis comes from studies that suggest a correlation between adhesiveness and clinical severity.
A variety of surface adhesion molecules on sickle erythrocytes are involved in this abnormal endothelial interaction. Sickle reticulocytes are especially prone to adhesion, having recently exited the marrow (where adhesion is required during erythroid maturation) and without any splenic filtrative circulation to allow “grooming” of the sticky young erythrocytes. Sickle erythrocytes bind to both the endothelial cells and to subendothelial extracellular matrix proteins such as laminin, thrombospondin, and fibronectin, which can become exposed after adhesion. Sickled cells have the highest affinity for laminin because of two isoforms of the protein that bear the Lutheran blood group antigens (B-CAM and LU). Adhesion proteins to thrombospondin on reticulocytes include CD47 and VLA-4 (α 4 β 1 integrin, which also bind to VCAM-1), whereas CD44 is the adhesion protein that binds to fibronectin.
Endothelial cells have different surface molecules depending on their location. For example, CD36 is expressed on microvascular but not large-vessel endothelium. Endothelial cells also express differing amounts of surface molecules and become more or less adherent depending on environmental stimuli. Cytokines such as interleukin-18 and tumor necrosis factor promote endothelial adherence, largely because of up-regulated VCAM-1 expression. In the setting of infection and inflammation, endothelial cells also modify their adhesion molecule profile.
Circulating endothelial cells can be identified in patients with SCA, and they exhibit an activated phenotype including surface expression of ICAM-1, VCAM-1, E-selectin, P-selectin, and tissue factor. Increased production of endothelial derived adhesion molecules (VCAM-1, ICAM-1, and E-selectin) was observed in 10 adult patients during acute SC pain crises. Soluble levels of VCAM-1, ICAM-1, and E-selectin were elevated and independently associated with organ damage, pulmonary hypertension (PHT), and mortality.
Membrane Lipid Orientation and Coagulation Defects
Normally phospholipids of the erythrocyte membrane are partitioned with amino phospholipids such as phosphatidylserine (PS) and phosphatidylethanolamine (PE) located on the inner cytoplasmic surface, whereas sphingomyelin and phosphatidyl choline (PC) are exposed on the outer surface (see Fig. 20-1 ). This asymmetry is not unique to the red cell and probably reflects a general structural pattern that keeps the amino phospholipids from activating soluble coagulation factors. In RSC, PS and PE flip back and forth from the inner leaflet to the outer leaflet during oxygenation and deoxygenation. Exposure of PS or PE on the outer surface during sickling may lead to increased phagocytosis by PS-binding proteins secreted by macrophages and may bind to specific PS receptors on endothelial cells. Alteration of the fatty acyl groups in PC results in changes in cell shape and deformability in SS cells, suggesting that the species composition of PC can affect membrane permeability and cellular deformability.
Abnormal lipid asymmetry accelerates clotting in vitro, and PS-positive sickle erythrocytes are correlated with markers of thrombin generation (prothrombin fragment F1.2, thrombin–antithrombin complexes) and fibrin degradation (D-dimers and plasmin–antiplasmin complex). Other markers of thrombophilia observed in patients with SCA are reduction in proteins C and S, platelet activation, and microparticles from endothelial cells, platelets, monocytes, and red cells that promote coagulation and may increase during crises.
Procoagulant and anticoagulant proteins as well as platelets play an important role in sickle erythrocyte adhesion to the endothelium. With sickle erythrocytes already having increased adhesiveness, the additional effects of increased levels of von Willebrand factor, activated platelets, and thrombospondin further increase RBC adhesiveness and make SCA a legitimate hypercoagulable state. Investigation with anticoagulants and antiplatelet agents are just now beginning, and may have a role in the prevention or amelioration of vaso-occlusive events.
Hemolysis and Nitric Oxide Depletion
The erythrocyte life span is shortened in SCA to approximately 10 to 20 days. The premature destruction of sickle erythrocytes includes both extravascular clearance by the RES (two thirds contribution) and intravascular processes (one third contribution). Extravascular hemolysis is enhanced by externalized PS exposure on the erythrocytes, which is recognized by macrophages with PS-specific receptors. The membrane changes found on sickle erythrocytes are similar to those seen in nucleated cells undergoing apoptosis (externalization of PS, annexin binding to and blebbing of the membrane, and cell shrinkage). Intravascular hemolysis is probably occurring at a steady-state (with 4 µM of plasma Hb) but with exacerbations caused by infectious or immunologic etiologies that lead to the concept of hyperhemolysis. In this setting, the massive release of Hb into the plasma overwhelms the normal binding and clearance mechanisms of haptoglobin and hemopexin and may lead to pathologic outcomes.
Nitric oxide (NO) is a critically important molecule that regulates vascular tone and functions as a cell-signaling molecule. NO is produced by a specific synthetase found in endothelial cells (endothelial nitric oxide synthase [eNOS]) that use L-arginine in an NADPH-dependent enzymatic reaction. NO increases intracellular cyclic guanosine monophosphate (GMP), which decreases smooth-muscle calcium concentrations, leading to muscle relaxation, vasodilation, and increased regional blood flow. NO also suppresses platelet aggregation, secretion of procoagulants, and reduces expression of endothelial cell adhesion molecules. Patients with SCA have elevated levels of plasma-free Hb, which consumes NO with resulting functional consequences of an “NO-depletion” state. Intravascular hemolysis has been postulated to cause a wide variety of clinical events in patients with SCA such as PHT, priapism, leg ulcers, stroke, and renal insufficiency, although the validity of this hypothesis has been recently called into question. Newer evidence suggests that an entirely different pathway may lead to PHT, one that involves marrow hyperplasia and high levels of circulating placental growth factor, which leads to endothelin-1 release and pulmonary vasoconstriction.
Acute and Chronic Inflammation
The HbS mutation and the pathophysiology of repeated sickling with erythrocyte–endothelium interactions leads to an acute and chronic inflammatory state with widespread endothelial dysfunction and vasculopathy. Markers of inflammation include increased levels of leukocytes, neutrophils, platelets, C-reactive protein (CRP), von Willebrand factor, and fibrinogen, all increased at steady state and during crises. Increased adhesion of sickle erythrocytes, reticulocytes, neutrophils, and platelets to vascular endothelium can initiate local inflammatory responses that alter the expression of endothelial surface molecules and lead to release of soluble forms into the plasma. Hypoxia, increased proinflammatory cytokines, and infections can activate vascular endothelium and leukocytes, which in turn can further increase adhesion molecules on both circulating cell surfaces and on endothelial cell surfaces (see Fig. 20-1 ). Over time, the endothelium becomes damaged enough that a chronic vasculopathy develops, causing clinical complications even without any acute sickling or vaso-occlusive events.
Circulating leukocytes and in particular neutrophils are also involved in the pathophysiology of SCA. Baseline white blood cell (WBC) and neutrophil counts in patients with SCA are elevated in the steady state, usually to twice the normal value. The degree of elevated WBCs is directly related to early mortality and increased acute chest syndrome (ACS) and has been suggested as a predictor of clinical severity in affected infants. Activated neutrophils are common in the steady state and also during vaso-occlusive crises, and they have enhanced adhesion to endothelium during vaso-occlusive events. In a transgenic sickle mouse model, neutrophils are involved in reperfusion inflammation after hypoxia, but the process is abrogated by blocking adherence with an antibody to P-selectin, which has led to therapeutic efforts in humans that target the selectins as a means to reduce vaso-occlusive pain. E-selectin has been identified as a crucial portal for invasive pneumococcal disease in a mouse model of SCA, and its down modulation by hydroxyurea can prevent fatal infection.
Recently two additional inflammatory pathways have been identified that have relevance for the pathophysiology and clinical manifestations of SCD. The first is the invariant natural killer T-lymphocyte (iNKT) cell, which when activated can mediate pulmonary inflammation and has been associated with the development of ACS. The adenosine A2A receptor (A2AR) agonist regadenoson reduces iNKT-cell activation and can decrease inflammation in sickle cell mice, and this agent is currently in human clinical trials. The second pathway is through mast cell activation, which promotes neurogenic inflammation and nociceptor activation in a murine model; specific inhibition with imatinib reduced hyperalgesia and suggests that pharmacologic manipulation of mast cells may influence inflammation and vaso-occlusive pain. General reduction of inflammation using statins may improve endothelial function as well. A pilot study demonstrated that simvastatin was safe, well-tolerated, and had benefits on markers of inflammation suggesting a potential therapeutic role.
Genetic Modifiers
In some ways SCA can be considered a simple disease, at least from the standpoint of pathogenesis, because the precise genetic mutation is known, the selection pressure for the origin and propagation of the sickle mutation is understood, the genetics of inheritance and transmission are straightforward, and the expression of the abnormal β S allele is predictable and deleterious. Despite this designation as a simple monogenic disorder, however, SCA is highly complex because of its phenotypic variability. Patients have a wide range of laboratory findings, clinical disease expression, and severity outcome that presumably reflect additional genetic and environmental influences.
Given the clarity of the pathogenetic basis for SCA, it is surprising how difficult the search for genetic modifiers of SCA has been. Perhaps because of differences in the definition or measurement of certain phenotypes, or because there are additional confounders, the identification of genetic modifiers for common laboratory and clinical events in SCA has been mostly fruitless, with a few notable exceptions. Even common mutations that originated in Africa with the same survival advantage against malaria do not have a predictable effect. For example G6PD deficiency is a common single-gene variant that also protects against malaria. In the United States, the prevalence of the A variant is about 10% to 13% among males, and there is no increased incidence of G6PD deficiency in SCD. Within the Cooperative Study of Sickle Cell Disease (CSSCD), the presence of G6PD deficiency in 801 males was not associated with more hemolysis or increased anemic episodes. Presumably no effects were observed because this mild variant of G6PD deficiency typically becomes manifest only in older erythrocytes, and the erythrocyte lifespan observed in SCA is very short. More recently G6PD deficiency has been associated with a risk of cerebral vasculopathy among children with SCA, although this association has not been verified by other investigators.
α-Thalassemia trait is another common finding among patients with SCA, with a prevalence of 30% to 40% in the United States and exceeding 50% in some parts of the world. The most common form of α-thalassemia trait is caused by a 3.7-kb deletion involving the entire α 2 gene. Additional deletional and nondeletional forms of α-thalassemia are found but are not as common. The primary effect of concomitant α-thalassemia is to limit the supply of α globins and thus further favor the competition of nonsickle β globin (γ globin) in the intracellular struggle for available α globins. This lowers the intracellular HbS content and thus prolongs the delay time for HbS polymerization. The resulting erythrocytes are more deformable because they have higher membrane area to volume ration; hence they have less potassium loss and cellular dehydration and a lower MCHC. Patients with SCA and α-thalassemia tend to have smaller, lighter cells with a higher circulating Hb concentration and lower rate of hemolysis. Indeed α-thalassemia trait combined with SCA has been associated with a lower risk of cerebral vasculopathy and stroke, a finding recently confirmed in a large cohort of children with SCA. However this combination does not necessarily translate into a better clinical outcome, because α-thalassemia has been associated with more vaso-occlusive pain, renal infarction, and avascular necrosis (AVN) of the femoral head. This may be the result of increased hematocrit and concomitant increase in viscosity in certain organs.
The baseline HbF levels in patients with SCA are highly variable, ranging from very low values (<5%) to very high values (>30%). Because HbF is known to ameliorate the phenotypic expression by reducing HbS polymerization, the origin of this marked variability has been of great interest. In the CSSCD any increment of HbF above 4% was associated with reduced painful events, and HbF levels greater than 8.6% were associated with decreased mortality.
Three broad categories of genetic mutations have been described that increase HbF postnatally. The first category includes large deletions of the γδβ-globin gene locus region on chromosome 11, which are rare but result either in δβ-thalassemia or pancellular hereditary persistence of fetal Hb (HPFH) as described in Chapter 21 . Patients who inherit HbS on one allele and a large pancellular HPFH deletion on the other have a condition that does not sickle under physiologic conditions. Patients with HbS/HPFH represent a unique syndrome with 20% to 35% HbF levels overall and distributed in all red cells, which results in normal Hb concentration, mild microcytosis, no sickled erythrocytes on the peripheral blood smear, and no clinical disease manifestations.
A second category of mutations that increase HbF in patients with SCA is linked to the β-globin gene region but does not feature large deletions of the gene locus and so has historically been termed nondeletional HPFH. HbF is usually distributed heterogeneously in these disorders, but because the fraction of F cells has a tight but nonlinear association with the %HbF, some of these patients approach a pancellular distribution. Despite the linkage to the β S mutation, a search within the promoter region of the γ-globin genes typically does not identify any specific mutations. The only nondeletional mutation clearly associated with an increased %HbF is the −158C to T polymorphism (rs7482644, also called the XmnI polymorphism because of the creation of a restriction enzyme cutting site) found in the G γ promoter, which is found in the Senegal and Arab-Indian (Saudi) β-globin haplotypes. These haplotypes are found commonly in genetic isolates in Western Africa, in Shiite Saudi Arabians, and in the Orissa region of India. Among patients in the United States with SCA, fewer than 10% have one chromosome with either of these haplotypes and fewer than 2% are homozygous for the Senegal haplotype.
The third category of genetic mutations that influence HbF levels in patients with SCA are those not linked to the β-globin region. Some patients have HbF levels ranging from very low (<3%) to much higher levels (15% to 20%); the latter are sometimes referred to as patients with HbSS/high HbF to distinguish them from HPFH variants. Many nonglobin genetic loci regulating HbF have been postulated and pursued including an elusive X-linked locus, but discovery of the BCL11A gene as a potent regulator of %HbF levels has vindicated this pursuit and provides mechanistic insights into HbF regulation and even therapeutic opportunities for patients with thalassemia and SCA. The BCL11A locus on chromosome 2p15 was first identified as a modifier of HbF levels using genome wide-association studies and subsequently this locus has been shown to have the greatest genetic influence on baseline HbF levels identified to date. The BCL11A protein is now recognized as a major biologic repressor of γ-globin gene expression, and silencing its expression can yield “de-repression” with high HbF levels and correction of the sickle phenotype in a murine model. In humans at least four single nucleotide polymorphisms affect BCL11A expression and thus HbF levels.
An additional gene locus regulating HbF levels on chromosome 6 has been pinpointed to the intergenic region of HBS1L-MYB at 6q23. Several noncoding polymorphisms within this region have been associated with higher HbF levels, but to date, no specific mechanisms have been elucidated for this genetic locus, although changes in MYB expression seem to be important. It is important to note that all genetic modifiers of HbF have not been identified; together, genetic variants within the BCL11A , HBS1L-MYB , and β-globin loci account for only 30% of the phenotypic variability of endogenous HbF expression and 50% of the heritable variability. Additional genetic regulators of HbF will be identified, but their effects will likely be smaller and their prevalence will be much lower.
Additional genetic modifiers that influence specific clinical phenotypes have long been sought for patients with SCA, including stroke, pain, ACS, leg ulcers, nephropathy, and AVN, among others. It is likely that networks of genes that respond to or modulate hemolysis, vasoregulation, inflammation, cell adhesion, hemostasis, and oxidative stress play roles in these phenotypes (see Fig. 20-1 ). Unfortunately most of these efforts have been hampered by the lack of accurate and rigorous phenotypes, small sample sizes, high false-discovery rates, and absence of validation cohorts. Moreover studies that investigate candidate genes are inherently limited to those genes, so unknown genes or networks can only be identified using unbiased “agnostic” approaches. Finally discovering an association between a genetic variant and a phenotype does not necessarily mean causality, and functional studies are needed to confirm the biologic significance and effect of any specific genetic association. The importance of a validation cohort is illustrated by the report that 90% of previously published genetic polymorphisms associated with stroke were not associated with stroke in a new cohort of children with documented stroke phenotype. At this time few genetic variants have been found that reliably influence any specific phenotype in patients with SCA except the UGT1A1 promoter polymorphism, which is also potent in the normal population for bilirubin levels. With improved gene-sequencing techniques including whole exome analysis, it is likely that new genetic variants will be identified that influence clinical phenotypes and even treatment responses to hydroxyurea.
Diagnosis
Fetus
The ability to perform prenatal diagnosis for SCD has progressed significantly since 1978 when Kan and Dozy described the DNA polymorphisms around the β-globin gene that were in linkage disequilibrium with the β S gene. Techniques for identification of the A-to-T substitution in codon 6 responsible for the glutamic acid-to-valine change in β globin have evolved from the use of restriction enzymes and homologous synthetic oligonucleotides to polymerase chain reaction–based amplification of DNA and sequencing. Sufficient DNA can be obtained from a small number of fetal cells obtained by amniocentesis or as early as 8 to 10 weeks’ gestation by chorionic villous biopsy. The desire to avoid the small risk of fetal loss associated with these invasive techniques has led to investigation of noninvasive prenatal diagnosis by testing fetal cells or free fetal DNA known to circulate in the plasma of pregnant women. Preimplantation genetic diagnosis (PGD) is now an established reproductive alternative to prenatal diagnosis in many specialized centers. Single-cell polymerase-chain reaction diagnosis of blastomeres followed by selection of unaffected embryos for standard in vitro fertilization has led to the birth of unaffected twins to a couple both with sickle trait. PGD has been used to facilitate selection of an unaffected histocompatible sibling donor for hematopoietic stem-cell transplantation of SCD.
Newborns
The newborn with SCD is generally not anemic or symptomatic until toward the end of the first year of life because of the protective effects of fetal Hb. Screening combined with comprehensive follow-up care was first begun by Pearson in New Haven and Serjeant in Jamaica in the 1970s. Because recognition of the disease in a newborn can lead to prevention of mortality and morbidity, the National Institutes of Health (NIH) recommends that all infants be screened in the neonatal period for SCD, and as of 2007 all 50 states and the District of Columbia perform universal screening for SCD. The method used for initial screening varies from state to state with most using isoelectric focusing (IEF) or high-performance liquid chromatography (HPLC) as their primary screening method. These tests can be performed on cord blood or on dried blood specimen blotted on filter paper. False negative screens using these methods have been reported in infants who received perinatal transfusions before screening. Diagnosis can also be performed using polymerase-chain reaction amplification of deoxyribonucleic acid (DNA) extracted from filter paper. Universal screening compared with targeted screening of newborns of parents at risk has been shown to identify more infants with disease, prevent more deaths; and is cost effective in areas where sickle trait is present in 7 to 15 of 1000 births. Considerations of logistics and equity have contributed to the decision for universal screening in the United States and the United Kingdom.
Older Children
After the first few months of life, as β S -globin production increases and HbF declines, the clinical syndrome of SCD emerges ( Table 20-1 ). Although at 1 week of age the Hb level of infants with HbSS is statistically lower than that of normal HbAA infants, the overlap between the two groups is considerable and does not diverge much before the second month of life. Anemia and reticulocytosis is usually evident by 4 months of age. ISCs are often absent from the peripheral blood of young children, and the morphology is typical of that of normal newborns: target cells, fragments, and poikilocytes. By 3 years of age, the typical peripheral blood smear is seen, including ISCs, target cells, spherocytes, fragments, biconcave disks, Howell-Jolly bodies, and nucleated red cells. The amount of HbF decreases with age, as in normal children, but this occurs much more slowly.
| Percentile | Age (mo) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-3 | 4-5 | 6-8 | 9-11 | 12-14 | 15-17 | 18-23 | 24-29 | 30-35 | 36-47 | 48-60 | ||
| SS Disease | ||||||||||||
| Hemoglobin level (g/dL) | 5 | 7.0 | 7.0 | 7.1 | 7.2 | 7.2 | 7.2 | 7.1 | 6.9 | 6.7 | 6.4 | 6.6 |
| 50 | 9.3 | 9.2 | 9.2 | 9.2 | 9.1 | 9.0 | 8.9 | 8.6 | 8.3 | 8.1 | 8.3 | |
| 95 | 11.4 | 11.3 | 11.4 | 11.5 | 11.5 | 11.5 | 11.3 | 11.1 | 10.9 | 10.5 | 10.4 | |
| Mean corpuscular volume (fL) | 5 | 72 | 69 | 68 | 67 | 67 | 67 | 67 | 68 | 69 | 71 | 72 |
| 50 | 84 | 81 | 95 | 96 | 94 | 94 | ||||||
| 95 | ||||||||||||
| Fetal hemoglobin level (%) | 5 | 14.6 | 12.3 | 10.8 | 9.1 | 7.8 | 6.7 | 5.6 | 4.8 | 4.5 | 4.4 | 3.3 |
| 50 | 43.5 | 34.1 | 29.1 | 24.3 | 20.6 | 17.7 | 14.8 | 12.8 | 12.4 | 12.4 | 9.0 | |
| 95 | 68.5 | 59.0 | 53.0 | 47.3 | 42.7 | 39.1 | 35.3 | 32.5 | 31.2 | 29.6 | 21.9 | |
| Reticulocyte count (%) | 5 | 1.0 | 1.1 | 1.2 | 1.3 | 1.4 | 1.6 | 1.9 | 2.3 | 2.6 | 2.7 | 1.8 |
| 50 | 4.0 | 5.1 | 5.9 | 6.7 | 7.4 | 8.0 | 8.7 | 9.3 | 9.8 | 10.4 | 11.8 | |
| 95 | 15.5 | 17.9 | 19.4 | 20.7 | 21.8 | 22.5 | 23.2 | 23.5 | 23.6 | 23.6 | 25.8 | |
| SC Disease | ||||||||||||
| Hemoglobin level (g/dL) | 5 | 8.0 | 8.2 | 8.6 | 8.9 | 9.2 | 9.3 | 9.5 | 9.5 | 9.4 | 9.3 | 9.6 |
| 50 | 9.7 | 9.8 | 10.1 | 10.3 | 10.5 | 10.6 | 10.7 | 10.8 | 10.7 | 10.6 | 10.6 | |
| 95 | 11.6 | 11.5 | 11.7 | 11.8 | 12.0 | 12.0 | 12.1 | 12.2 | 12.2 | 12.1 | 11.9 | |
| Mean corpuscular volume (fL) | 5 | 68 | 65 | 64 | 64 | 63 | 63 | 63 | 63 | 64 | 66 | 69 |
| 50 | 81 | 78 | 77 | 75 | 74 | 74 | 74 | 74 | 76 | 77 | 77 | |
| 95 | 91 | 88 | 86 | 85 | 84 | 84 | 84 | 84 | 86 | 88 | 87 | |
| Fetal hemoglobin level (%) | 5 | 13.6 | 2.9 | 2.9 | 3.1 | 3.1 | 2.9 | 2.4 | 1.4 | 0.5 | 0.0 | 2.0 |
| 50 | 31.6 | 17.9 | 14.5 | 11.6 | 9.3 | 7.4 | 5.5 | 4.2 | 3.9 | 4.4 | 4.2 | |
| 95 | 54.0 | 39.1 | 32.1 | 25.7 | 20.9 | 17.6 | 14.7 | 13.8 | 14.7 | 15.9 | 8.3 | |
| Reticulocyte count (%) | 5 | 0.8 | 0.8 | 0.8 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.9 |
| 50 | 2.8 | 2.8 | 2.7 | 2.6 | 2.5 | 2.5 | 2.5 | 2.6 | 2.7 | 2.9 | 2.8 | |
| 95 | 8.2 | 8.8 | 8.9 | 9.0 | 8.9 | 8.7 | 8.4 | 8.0 | 7.9 | 8.8 | 13.4 | |
| S β + -Thalassemia | ||||||||||||
| Hemoglobin level (g/dL) | 5 | 9.2 | 9.4 | 9.1 | 8.5 | 9.1 | 9.1 | 9.9 | 10.0 | 9.8 | 9.3 | 10.0 |
| 50 | 10.8 | 10.9 | 11.0 | 10.8 | 10.6 | 11.2 | 10.9 | 11.0 | 10.7 | 10.6 | 10.8 | |
| 95 | 12.4 | 12.7 | 13.5 | 11.8 | 14.1 | 12.0 | 12.0 | 13.0 | 11.3 | 11.6 | 11.2 | |
| Mean corpuscular volume (fL) | 5 | 70 | 64 | 61 | 61 | 63 | 82 | 63 | 61 | 66 | 64 | 66 |
| 50 | 80 | 73 | 72 | 69 | 72 | 70 | 70 | 70 | 72 | 76 | 68 | |
| 95 | 88 | 83 | 84 | 75 | 84 | 73 | 77 | 79 | 76 | 76 | 76 | |
| Reticulocyte count (%) | 5 | 1.1 | 0.0 | 1.1 | 0.9 | 0.8 | 0.9 | 0.7 | 1.5 | 1.2 | 1.0 | 3.0 |
| 50 | 2.6 | 1.8 | 2.5 | 2.5 | 3.0 | 2.5 | 2.4 | 2.2 | 3.4 | 2.2 | 4.1 | |
| 95 | 8.5 | 6.4 | 2.5 | 4.6 | 5.9 | 7.4 | 5.1 | 6.2 | 7.4 | 7.6 | 5.7 | |
Clinical Manifestations
The clinical manifestations of SCD are extremely variable. Some patients are entirely asymptomatic, whereas others suffer often from acute painful episodes. Most patients fall between these extremes and have relatively long asymptomatic periods punctuated by occasional clinical crises. The complex nature of the clinical variability from patient to patient and over time in each patient has been prospectively studied on a large scale in the CSSCD, under the auspices of the Sickle Cell Disease Branch of the National Heart, Lung, and Blood Institute (NHLBI).
Acute Sickle Cell Crises
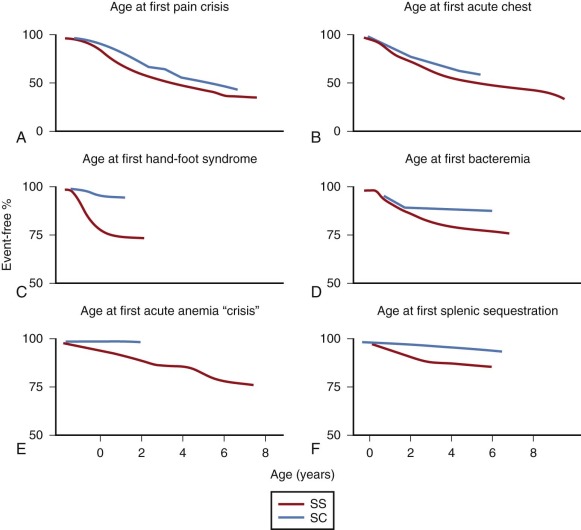
The term sickle cell crisis was defined by Diggs as “any new syndrome that develops rapidly in patients with SCD as a result of the inherited abnormality.” There are three categories of sickle crisis: vaso-occlusive, sequestration, and aplastic, and they are covered individually in the next few sections. The best perspective on how these acute events play out in a typical group of children comes from a report by Gill and colleagues describing the experience of almost 700 infants studied for 10 years as part of the CSSCD. The age at first event is displayed in Figure 20-6 .
Vaso-occlusive crises are acute, often painful episodes resulting from intravascular sickling and tissue infarction. In a prospective study of children with HbSS disease studied since birth in Jamaica, painful crisis was the first symptom in more than one fourth of the patients and the most common symptom after the age of 2 years. Vaso-occlusive crises (also called vaso-occlusive episodes or events ) are the major clinical manifestations of SCD and occur most commonly in the bones, soft tissues, lungs, liver, spleen, brain, and penis. Sometimes the differential diagnosis is difficult, because there is no definitive objective hallmark of vaso-occlusive crises and they can mimic other pathophysiology.
Acute Painful Crisis
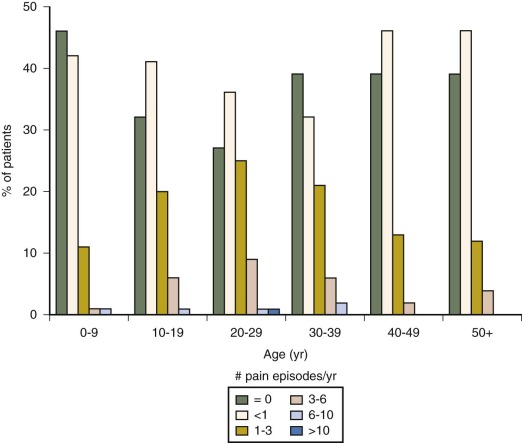
The most common acute vaso-occlusive crisis is pain. Virtually all patients with HbSS disease experience some degree of acute pain. For many these episodes are mild and managed entirely at home, school, or work. Little is known about the extent or nature of pain-coping activities that go on outside the medical setting, but diary studies suggest that they are numerous. The “tip” of the pain “iceberg” is made of those episodes that drive patients to seek medical attention. These episodes vary widely among patients ( Fig. 20-7 ) and represent the most common reason for patients to visit outpatient offices and emergency departments and be admitted for inpatient care. Despite the fact that there is variation in how or why individual patients decide to seek attention for a given episode of pain, epidemiologic evidence strongly indicates that patients with higher rates of medical attention for pain have lower levels of fetal Hb, higher WBC counts, higher steady-state Hbs, and higher mortality. Some studies have suggested that infections, changes in climate, and psychological factors may precipitate pain episodes, although commonly no precipitating factors can be identified.
Patients typically experience rapid onset of deep, gnawing, throbbing pain, usually without any abnormal physical or laboratory findings but sometimes accompanied by local tenderness, erythema, warmth, and swelling. The underlying pathology is bone marrow ischemia, sometimes leading to frank infarction with acute inflammatory infiltrate. The most commonly involved areas are the lumbosacral spine, knee, shoulder, elbow, and femur. Less often, the sternum, ribs, clavicles, calcaneus, iliac crest, mandible, zygoma, and mandible are involved. Sympathetic joint effusions during acute periarticular episodes are particularly common in the knees and elbows. Typically joint aspiration yields straw-colored fluid, usually with a “noninflammatory” profile. Rarely sterile purulent exudates are found. Given the range of marrow involvement and inflammatory response, it is not surprising that the findings in patients with the most inflammation mimic the findings of osteomyelitis, and those without such findings are at risk of being considered malingerers.
Even in patients with measurable signs of inflammation, the diagnosis of infarction is favored over osteomyelitis. The results of one study suggest that acute long-bone infarction is at least 50 times more common than osteomyelitis. In this study of 41 acute long-bone infarcts, 38% affected the humerus, 23% affected the tibia, and 19% affected the femur. All patients experienced local tenderness, with swelling in 85%, joint findings in 68%, and local heat in 65%. Fourteen percent appeared “toxic,” 21% had a temperature greater than 39° C, and 43% had a temperature lower than 38° C. The total WBC count ranged between 7200 and 43,000 cells/mm 3 , with a mean leukocytosis of 17,000 cells/mm 3 . The mean sedimentation rate was 30.5 mm/h, with a range of 3 to 66 mm/h. Although various radionuclide scans have been suggested to distinguish between infarction and infection, in many studies such investigations were inconclusive. Magnetic resonance imaging (MRI) of patients with HbSS disease shows decreased intensity of short relaxation time/echo-time pulse-sequence imaging, owing to hyperplastic marrow that converts to high intensity on long relaxation time/echo time images in painful crises, but no definitive series compares infarction to infection. Therefore despite the progress made in the development and use of imaging techniques, a definitive diagnosis of osteomyelitis in SCD still relies more upon clinical assessment together with positive cultures from blood or bone obtained by aspiration or biopsy than upon any single imaging modality.
Except for a positive blood or tissue culture, no laboratory test can differentiate acute infection from painful crisis. Needle aspiration and culture of the highly suspicious area is critical for isolating the organism and should be considered before initiating empirical antibiotic therapy. The aspirated fluid may be quite purulent even in the patients with sterile infarcts. In most series, the most common organism causing osteomyelitis is Salmonella though Staphylococcus , S. pneumoniae and Gram-negative enteric bacilli are also common. Initial empiric antibiotic therapy should be chosen to cover these possibilities. Treatment failures are seen when anything but the most aggressive antibiotic regimens are used.
As described earlier, episodes of acute bone pain and impressive signs of inflammation may be difficult to distinguish at outset from osteomyelitis. More common and just as challenging is the evaluation and management of severely painful episodes in patients without obvious signs of inflammation. Some of these patients show laboratory evidence of acute inflammation such as elevated CRP, fibrinolysis such as elevated D-dimers, or red-cell trapping such as loss of dense cells. These measurements are not helpful in the management of individual cases, and should not be used as an attempt to “validate” an individual patient’s report of symptoms. In a research setting, however, these measurements done on large numbers of patients with and without symptoms provide clues to potential innovative therapeutic interventions. For example the common finding of elevated acute-phase reactants stimulated a trial of methylprednisolone for treating acute bone pain. This preliminary work showed that a short course of high-dose corticosteroid decreased the duration of severe pain; however, it resulted in more “rebound” attacks after treatment was discontinued. Similarly despite the fact that previous trials of aspirin therapy were not encouraging, there is significant interest in the role of platelets, soluble procoagulants, anticoagulants, and endothelial cells in the precipitation or propagation of vaso-occlusion and has reopened these as potential lines of treatment.
In children younger than 5 years of age, the small bones of the hands and feet are often affected, even to the distal phalanges, and in contrast to most episodes of bone pain in older children, physical findings are common. This painful dactylitis (“hand-foot syndrome”) is typically the first clinical manifestation of SCD. The young child cries with pain; refuses to bear weight; and has puffy, tender, and warm feet, hands, or both. The child may appear acutely ill, be febrile, and have an impressive leukocytosis. At the onset of soft-tissue swelling, there are usually no bony changes on radiographs. After 1 to 2 weeks, subperiosteal new bone, irregular areas of radiolucency, cortical thinning, or complete destruction of bones can be seen. All the bone changes are usually reversible but may persist for as long as 8 months. A rare complication, permanent shortening of the digits after hand-foot crisis, has been reported. Dactylitis before age 1 year is a strong predictor of overall severity (stroke, death, high pain rate, or recurrent ACS) by 10 years of age, although recent single-institution evidence suggests dactylitis is not a strong predictor of subsequent pain or ACS. In addition to dactylitis the risk for severe disease is further increased if the child has also experienced an episode in which the Hb dropped below 7 g/dL, and/or the baseline WBC count was elevated.
Acute Chest Syndrome
ACS is an acute illness of lung injury characterized by chest pain, fever, and/or respiratory symptoms, accompanied by a new pulmonary infiltrate on chest radiograph. The diagnostic criteria most commonly include radiographic evidence of a new segmental pulmonary infiltrate and one or more of the following: fever, tachypnea, cough, or new-onset hypoxia; increased work of breathing (intercostal retractions, nasal flaring, accessory muscle use); or chest pain. The term acute chest syndrome was introduced by Charache et al and reflects the difficulty of establishing a definitive etiology and emphasizes the fact that in individual patients, knowing the specific etiology is less critical for management than being able to assess the magnitude and pace of the lung injury.
The critical pathophysiology is that only deoxygenated HbS polymerizes and that reoxygenation eradicates the polymer. The lung is the critical organ that protects the arterial side of the circulation from the sludge of sickle polymers. When the lung is injured, underventilated, or inflamed such that a ventilation/perfusion (V/Q ) mismatch exists, this protection is inadequate, and downstream tissues including the injured lung itself become increasingly susceptible to sickling and ischemia. In fact, when the flow is eventually restored in a sickle mouse model, reperfusion injury occurs with activation of iNKT cells and p-selectin–inhibitable movement of inflammatory cells into the tissue.
Although some patients, particularly adults, initially can provide a full-blown picture of chest pain, hypoxia, and abnormal x-ray findings, the diagnosis of ACS in young patients can be difficult to make. Often the diagnosis becomes clear only days into an event that started as a fever without source or pain. Children younger than 4 years of age often have few signs at presentation: in one study, 35% to 40% had a normal lung examination; 30% to 40% had no tachypnea; and 30% to 50% had no tachycardia. The radiographic evolution often lags behind the clinical progression. In one series, only 36% of patients had an abnormal x-ray on presentation, although all eventually developed abnormal radiographic findings.
Vichinsky and associates demonstrated that almost half of all patients who developed ACS were initially admitted for other reasons, often with vasoocclusive pain. Opioid analgesics and pain of the spine, ribs, and abdomen can lead to hypoventilation with decreased tidal volume, development of atelectasis, and V/Q mismatch. The subsequent regional hypoxemia results in further intrapulmonary sickling. Secretory phospholipase A2 (sPLA2) and serum CRP have been proposed as possible laboratory predictors of ACS development but not widely adopted. Elevated sPLA2 as a trigger for simple transfusion appeared to reduce the risk of developing ACS compared with standard therapy in one small uncontrolled trial, but a larger multicenter feasibility trial was challenged by poor enrollment.
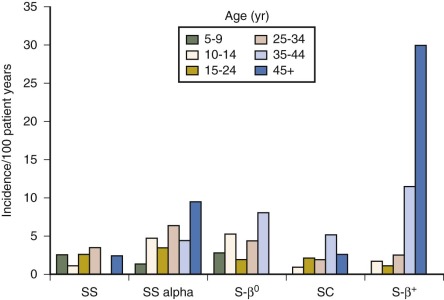
ACS is a leading cause of morbidity and mortality in SCD. It represents the second most common acute complication (pain episode is first) with a rate of 12.8 cases per 100 patient-years and the most common condition at the time of death. The highest incidence is in children aged 2 to 4 years (25.3 per 100 patient-years in SCA), and lowest in adults (8.8 per 100 patient-years in SCA). Patients who have not experienced an episode of ACS have a longer life expectancy than those who have, and those with a higher level of HbF and lower steady-state WBCs have a lower attack rate for ACS. Induction of fetal Hb with hydroxyurea therapy lowers incidence of ACS in all age groups.
Asthma or reactive airways may represent a distinct or overlapping comorbid condition and appears to be a risk factor for vaso-occlusion and for ACS. Patients with SCD and asthma were two to four times more likely to develop ACS than those without asthma, had longer hospital stays, and experienced a higher incidence of painful episodes. Analysis of the CSSCD cohort revealed that comorbid asthma is a significant risk factor for mortality for patients with SCD and should be treated aggressively.
Vichinsky and colleagues prospectively studied 671 episodes of ACS. The most common etiologies were fat/bone-marrow embolus, and bacterial, atypical bacterial ( Chlamydia, Mycoplasma ), viral, or mixed infection. In contrast to patients with infection, those with bone-marrow embolus were more likely to have an associated episode of bone pain, chest pain, abnormal neurologic symptoms, and fall in platelet count. Regardless of etiology ACS is always accompanied by some degree of localized sickling and lung ischemia. There is considerable change in blood counts at the time of presentation in patients with ACS. The Hb concentration drops an average of 0.7 g/dL that likely reflects sludging and intrapulmonary sickling, and WBC counts increase an average of 69%. The mean partial pressure of oxygen (pO 2 ) was 71 mm Hg with one fifth of the patients having a pO 2 less than 60 mm Hg. Transcutaneous SpO 2 measurements may not be reliable in SCD, and most clinical transcutaneous pulse oximeters overestimate the fractional arterial oxygenated (oxy-)Hb saturation. It is unwise, therefore, to place too much reliance on single SpO 2 measurement in isolation.
Several important therapeutic principles emerged from the Vichinsky study. First, bronchodilator therapy was remarkably effective in patients with wheezing or pulmonary function tests indicating obstruction. This is consistent with the observation that measurable airway hyperactivity exists in many children with HbSS even without a history of asthma. Second, red-cell transfusion improved oxygenation. Simple transfusion is as effective as exchange transfusion and may indicate that rapid reversal of intrapulmonary sickling is therapeutic. Third, the prominence of Chlamydia and Mycoplasma underscores the importance of including macrolide antibiotics in the initial broad empiric therapy.
All patients with ACS must be managed in the hospital setting with frequent and close monitoring. Broad spectrum antibiotics (including a macrolide), bronchodilator therapy, frequent incentive spirometry, and simple transfusion represent the mainstay of early intervention. Oxygen therapy should be provided for patients with hypoxia, and exchange transfusion is indicated for worsening hypoxia or clinical deterioration despite simple transfusion. Chest and rib pain often lead to splinting and atelectasis that may exacerbate or even precipitate ACS; therefore analgesic therapy should not be withheld but should be titrated to analgesic effect with careful monitoring and frequent reevaluation. Fluid overload with pulmonary edema/effusion and congestive heart failure can occur, so careful attention to fluid balance is critical. For patients with poor respiratory effort or rising oxygen requirements, the use of noninvasive ventilation such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BiPAP) may be useful. Progression to respiratory failure is not uncommon, and if the patient is ventilated, bronchoscopy may have both diagnostic and therapeutic benefit. Detection of plastic bronchitis and removal of branching bronchial casts can improve hypoxemia and V/Q mismatch. Although most patients with ACS recover uneventfully, the course is unpredictable and can progress to rapid, unexpected deterioration that leads to multiorgan failure and death.
Acute Abdominal Pain
Severe acute abdominal pain is a common presentation that can pose a difficult differential diagnosis. The etiology of abdominal pain in children with SCD is often benign (e.g., secondary to constipation or gastroenteritis) but can reflect mesenteric sickling and vertebral disease with nerve root compression. A true sickle-related abdominal crisis can be accompanied by guarding, tenderness, rebound, fever, and leukocytosis that is indistinguishable from an acute surgical abdomen. Often the patient is the best judge and can state whether the pain is characteristic of a sickle “crisis.”
These patients should receive general supportive measures with the omission of high-dose opioid analgesics until the clinical trajectory becomes clear. Patients with severe pain should be given nothing by mouth and followed closely by both medical and surgical personnel. Abdominal films, including upright views, may be helpful in identifying retained stool, air-fluid levels consistent with infection-related hypomotility, or even a perforated viscus. Usually the patient with vaso-occlusive pain remains stable or improves slightly with hydration and mild sedation. In extreme cases in which the clinical situation is deteriorating, emergency surgical exploration may be necessary. Simple or exchange transfusion should be done before surgery if possible.
Acute focal right upper quadrant pain may be a result of acute cholecystitis or intrahepatic sickling. Abdominal ultrasonography will indentify cholelithiasis and choledocholithiasis in the hands of a skilled operator. The presence of gallstones and a clinical syndrome characteristic of cholecystitis is common but does not always mean that the symptoms are the result of the stones. However most gallstones in young patients with SCD are symptomatic, and patients benefit from elective cholecystectomy. Patients with acute cholecystitis should have surgery only after the gallbladder has had a chance to “cool down,” because perioperative management of emergent surgery can be fraught with complications. (For further discussion see the Hepatobiliary System section).
On rare occasions there may be intrahepatic sickling and even hepatic sequestration. Intrahepatic sickling can result in acute right upper quadrant pain from acute sickle hepatic crisis, hepatic sequestration, and/or SCD intrahepatic cholestasis. Acute hepatic crisis commonly presents with acute right upper quadrant pain, tender hepatomegaly, nausea, low-grade fever, and jaundice. Transaminases are rarely greater than 300 IU/L and serum bilirubin levels are usually less than 15 mg/dL. The crisis usually resolves within 2 weeks with IV hydration and analgesia. Hepatic sequestration also presents with sudden tender hepatomegaly, but in contrast presents with an acute anemia and without cholestasis or transaminitis. Treatment is to replace circulating volume with crystalloid or simple transfusion. Transfusion must be undertaken judiciously, because the liver may “release” the endogenous sickle erythrocytes leading to elevated hematocrit consequent problems secondary to hyperviscosity. Exchange transfusion should be considered. SCD intrahepatic cholestasis is a syndrome of right upper quadrant pain, nausea/vomiting, fever, tender hepatomegaly, leukocytosis, and extreme direct hyperbilirubinemia. There is a broad spectrum of severity in intrahepatic cholestasis that can include acute liver failure with bleeding diathesis, encephalopathy, renal failure, and progression to multiorgan failure and is associated with high mortality, particularly in adults. These hepatic complications are not to be confused with the “benign hyperbilirubinema” described by Buchanan et al and is characterized by marked conjugated hyperbilirubinemia, only mild transaminitis, and no liver synthetic dysfunction. This entity resolves spontaneously within 2 to 8 weeks and may represent a very mild variant of intrahepatic cholestasis.
Acute Central Nervous System Event
Acute infarction of the brain can result in a devastating stroke, which occurred in approximately 7% to 10% of children with SCA in the era before transcranial Doppler (TCD) screening. The incidence is estimated to be 0.7% per year during the first 20 years of life, with the highest rates in children 5 to 10 years of age. Stroke may occur as an isolated event but also appears in the setting of evolving ACS, aplastic crisis with severe anemia, viral illness, painful crisis, priapism, or dehydration. The most common underlying lesion is an intracranial arterial stenosis or obstruction, usually in the distal internal carotid, the proximal middle cerebral, or the anterior cerebral arteries. Pathologically these vessels whose endothelium has presumably been chronically injured by sickle erythrocytes show hyperplastic intima with proliferation of fibroblasts and smooth muscle. The lumen may be narrowed or completely obliterated by the vascular lesion, suggesting that acute sickling may simply be the “last straw,” causing acute infarction in the setting of a chronic vasculopathy. Hemiparesis, focal seizures, gait dysfunction, and speech defects are the most common presenting signs, but persistent cognitive and psychological impairments commonly accompany motor deficits after stroke.
A careful discussion of the evaluation of the child with new neurologic symptoms is provided by Adams. As soon as cerebral ischemia is suspected and the patient stabilized, the best initial diagnostic test is a noncontrast CT scan. However diagnostic imaging should never delay immediate therapeutic intervention. Although CT may not be positive for infarction within the first 6 hours of ischemia, it can rule out hemorrhage or nonischemic etiologies. MRI with diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences are highly sensitive and provide better detail of the areas of ischemia within minutes of ischemia and remain abnormal for several days. The typical findings are infarcts associated with major vessel obstruction or distal obstruction of smaller vessels leading to infarction in the “border zone” area between the anterior cerebral and middle cerebral vessels. Assessment of the intracranial and cervical artery vasculature using three-dimensional time-of-flight (TOF) magnetic resonance angiography (MRA) is becoming increasingly useful in the early evaluation of the patient with new symptoms and is nearly as accurate as conventional cerebral angiography.
Patients with suspected acute neurologic deficits should immediately receive IV hydration while blood is being cross-matched, because signs and symptoms of stroke can often be quickly reversed, even with isotonic IV fluids. Simple transfusion to a Hb target of approximately 10 g/dL is also helpful to alleviate acute intracranial sickling and can be accomplished in most clinical settings. In a tertiary hospital the standard approach to treating a patient with acute infarction is immediate exchange transfusion (see “ Transfusion ” for details). There are no data for the use of recombinant tissue plasminogen activator (tPA) in sickle-associated cerebral ischemia for either children or young adults.
Patients treated with simple or exchange transfusion usually show marked improvement in motor function, although the prognosis is considerably worse for those with multiple infarcts. After the initial transfusions, a chronic maintenance transfusion program with a goal of suppressing HbS to less than 30% reduces the incidence of stroke recurrence to 10% to 15%, representing a substantial reduction in stroke rate compared to no intervention. Patients whose initial stroke was temporally unrelated to another medical event (e.g., ACS) may be at higher risk for recurrent stroke even while receiving regular transfusions. The appropriate duration of chronic erythrocyte transfusions for secondary stroke prophylaxis has not been firmly established. Two prospective studies documented a high risk of recurrent stroke (60% to 70%) after discontinuation of either a short-term (1 to 2 years) or long-term (5 to 12 years) transfusion regimen. Therefore an indefinite transfusion program is currently recommended to prevent secondary stroke. Although clearly beneficial, indefinite maintenance transfusion carries the risks of allosensitization, autoantibody formation, transfusion-borne infections, and the certain progression to iron overload. Bone-marrow transplantation in patients who have had strokes can be curative, although parenchymal brain disease may progress.
Hydroxyurea treatment with increased HbF served as an alternative to chronic transfusion for effectively preventing secondary stroke in a single institution. The subsequent multicenter trial (Stroke With Transfusions Changing to Hydroxyurea study; SWiTCH) compared the efficacy of standard treatment (chronic transfusions and chelation) to alternative treatment (hydroxyurea and phlebotomy) for children with SCA, previous stroke, and iron overload. SWiTCH was terminated early after a scheduled interim analysis indicated statistical futility for reaching the composite primary study endpoint of both recurrent stroke rate and improved iron burden. The alternative treatment arm had an increased stroke recurrence rate compared with the standard treatment arm but within the predicted stroke margin of the study design; however, the iron unloading from serial phlebotomy was not superior to daily deferasirox, so the composite study endpoint was not reached. For the children in SWiTCH who had a recurrent stroke, risk factors included severe cerebral vasculopathy, younger age at first stroke, and history of transient ischemic attack (TIA). The duration of follow-up study may have been too short to demonstrate any differences in cerebral vasculopathy between the transfusion and hydroxyurea arms. Because of the inferior results on SWiTCH’s hydroxyurea and phlebotomy arm, chronic transfusion therapy with chelation remains the mainstay of secondary stroke prevention in children with SCA.
Patients who develop severe cerebral vasculopathy (moyamoya syndrome) appear to have a high risk for recurrent stroke despite adequate chronic transfusion. One retrospective evaluation of chronic transfusions for secondary stroke prevention revealed recurrent stroke was more common among patients with moyamoya (11/19 patients, 58%) than without moyamoya (7/25 patients, 28%). Given this increased risk, some advocate for surgical revascularization to restore circulation to the at-risk hemisphere. Although there are no randomized, controlled trials in children with SCA and moyamoya disease to determine the effectiveness of indirect surgical revascularization procedures such as encephaloduroarteriosynangiosis (EDAS) or pial synangiosis, postsurgical angiography has shown promising revascularization of the affected area.
Given the devastating consequences of overt clinical stroke and the difficulties in preventing recurrent stroke, application of radiologic techniques for noninvasive assessment of the brain and its vasculature has focused attention on two areas: identifying children at high risk for primary stroke by TCD ultrasonography and identifying children with asymptomatic “silent” cerebral infarcts (SCI) by MRI.
In the early 1980s the utility of TCD to measure intracranial arterial blood flow was first reported. TCD allows an easy, noninvasive method of measuring cerebral blood velocity in the circle of Willis. The highest time-averaged maximum velocity (TAMV) measured in the distal internal carotid artery (ICA) and proximal middle cerebral artery (MCA) has become the standard reported TCD value. In children with SCA, TCD can identify intracranial arterial vasculopathy, although an inverse relationship between hematocrit and TAMV values has been recognized. A landmark prospective single-institution study of untreated children with SCA demonstrated that higher TCD velocities in the distal ICA or proximal MCA were associated with an elevated risk of primary ischemic stroke. A subsequent study with a longer follow-up range confirmed that TCD velocities reflect a continuum of stroke risk, but 200 cm/s represents a potential threshold value, above which the risk of primary stroke is very high. For 315 untreated children with SCA studies for an average of 64 ± 24 months, 25 (8%) had TAMV greater than or equal to 200 cm/s, and 10 of these children (40%) had a primary stroke.
Similar to secondary stroke prevention with chronic transfusion, positive results have also been demonstrated for primary stroke prevention in SCA for children with abnormal TCD velocity. In the multicenter prospective Stroke Prevention Trial in Sickle Cell Anemia (STOP) with 130 asymptomatic children with abnormal TCD measurements (>200 cm/s), subjects were randomized to transfusions or standard care. The study was terminated early when only 1 of 63 transfused children compared with 11 of 67 untreated children had strokes, representing a 92% decrease in the relative stroke rate in the treated group. A subsequent randomized controlled trial (STOP 2) tested whether chronic transfusions for primary stroke prevention based on abnormal TCD could be safely discontinued after at least 30 months in children who had not had an overt stroke and who had reversion to low-risk TCD (<170 cm/s) with chronic transfusion therapy. After 79 of a planned 100 children were randomized, the trial was also terminated early because 16 of 41 subjects randomized to stop transfusion achieved endpoint (14 reversions to high-risk TCD without stroke and 2 ischemic strokes) versus 0 of 38 maintained on chronic transfusion treatment. The current TCD with Transfusions Changing to Hydroxyurea (TWiTCH) clinical trial is testing whether hydroxyurea can be used as an alternative treatment to indefinite transfusions in the setting of abnormal TCD.
A systematic study of MRI in asymptomatic children with SCA revealed that unanticipated SCIs are associated with an increased risk of subsequent clinical stroke. However in older children (mean age, 11 years) without evidence of prior central nervous system (CNS) disease, TCD and MRI results are often discordant, indicating that these tests may be measuring different aspects of CNS pathology. Brain MRI has revealed that 18% to 25% of children in the CSSCD had evidence of SCI. These silent infarcts are associated with impaired performance on psychometric tests that measure fine motor skills and cognitive ability. Older children (>6 years) with silent infarcts were more likely to have a history of seizures, low Hb level, and high WBC count (>11.8 × 10 9 /L). In a study of 392 children studied prospectively up to 10 years, elevated WBC count, dactylits, and low Hb (<7 g/dL) before the age of 2 years were associated with adverse outcomes including strokes. In other studies low hematocrit, ACS and hypertension have been associated with silent infarcts and/or clinical strokes. These observations suggest that MRI might be used to screen older children with poor school performance or evidence of other risk factors.
Clinical trials are underway for treatments of younger children who develop evidence on MRI of silent infarcts or abnormal intracranial Doppler flow measurements including transfusions, hydroxyurea, or bone marrow transplant.
Not all acute focal neurologic events in patients with SCA are infarcts. Intracranial hemorrhage is the other major category of sickle-related neurologic event, although the incidence is higher among adults than children. Hemorrhagic stroke presents as a sudden severe headache, sometimes with neck pain, vertigo, syncope, nystagmus, ptosis, meningismus, nausea/vomiting, or photophobia. Many of these episodes are subarachnoid hemorrhages from small bleeding aneurysms that probably arise from intimal damage during childhood. Multiple aneurysms with extensive collaterals (similar to moyamoya disease) are often found during angiographic evaluation of children with cerebrovascular disease. Successful aggressive neurosurgical approaches to these bleeding lesions have been published and suggest that angiography should be done to identify patients with surgically amenable lesions.
Priapism
Priapism occurs in males of all ages, but the incidence and prevalence are not well established. In a Jamaican study of 104 males age 10 to 62 years, 42% reported at least one episode of priapism, with a median age of onset of 21 years. The long-term follow-up study of patients in Los Angeles determined that approximately 7% of males with SS and about 2% of those with SC develop priapism, with a median age of onset of about 22 years. A questionnaire survey administered to 98 patients below the age of 20 in Dallas found the mean age at the initial episode to be 12 years, with an average number of episodes per patient of 15.7 and an 89% actuarial probability of experiencing priapism by age 20. Prolonged sexual arousal, fever, cold exposure, nocturnal tumescence (rapid eye movement [REM] sleep), full bladder, dehydration, alcohol, cocaine, and testosterone therapy have all been implicated as triggers. Because early intervention and treatment may prevent irreversible penile fibrosis and impotence, patients and parents should be educated in advance of its occurrence to prevent embarrassment and to treat prolonged priapism as a urological emergency.
Four clinical entities are described: stuttering priapism, with short (less than 2 to 3 hours), often multiple episodes; acute prolonged priapism (more than 24 hours), which can persist for weeks; chronic priapism, a painless induration that may last for years; and acute-on-chronic priapism, acute painful episodes complicating chronic induration. Sexual dysfunction was reported by 46% of patients with a history of priapism. In general young patients with brief episodes restricted to the corpora cavernosa (bicorporal priapism) are less likely to become dysfunctional, whereas adults with involvement of the corpora cavernosa and corpus spongiosa (tricorporal priapism) tend to have prolonged episodes and higher than a 50% chance of becoming impotent. Urinary retention requring catheterization may complicate such acute episodes. Patients with tricorporal involvement are also particularly susceptible to acute severe neurologic complications and overall increased morbidity, even those treated with exchange transfusion. They should be monitored closely for early neurologic symptoms, especially headache.
Diagnostic studies such as technetium scans, MRI, Doppler flow studies, and measurements of corporal pressures, Hb electrophoresis, and blood gases have been used in a variety of settings to help define the anatomy and potential prognostic features of priapism. As yet they have not had an impact on clinical decision making in individual patients.
A consensus on treatment of priapism has not been reached, and there are no controlled clinical trials to guide therapy. Short episodes of acute priapism usually occur on awakening and can be managed at home. Patients should be instructed to urinate often, perform mild exercise, increase oral fluid intake, and soak in a warm tub and take analgesics. If the episode does not resolve within a few hours, the patient should receive medical treatment for a typical vaso-occlusive episode with aggressive IV fluids and analgesics. If the episode persists beyond 4 hours, intracavernosal aspiration and instillation of an α agonist should be performed and repeated. Although the role of RBC transfusion is somewhat controversial, simple red-cell transfusion may relieve priapism and provide safety for urgent surgical intervention. Exchange transfusion may produce results in 6 to 8 hours, although most do not show signs of detumescence for 24 to 48 hours, and some do not respond at all. Use of intracorporal and oral α-adrenergic agents has been tried successfully in a small experimental group in Paris in both acute and chronic settings, and in one group of patients with stuttering priapism in Jamaica, stilbestrol was helpful in preventing recurrences. Numerous medical therapeutic options have been attempted, including diethylstilbestrol, gonadotropin-releasing hormone analogs, various adrenergic agonists, antiandrogens, baclofen, gabapentin, phosphodiesterase type 5 (PDE5) inhibitors, and hydroxyurea.
Surgical shunting procedures should be reserved for patients who have not shown any signs of detumescence 12 to 24 hours after corporal irrigation and/or transfusion, particularly if they are postpubertal. Even partial responses should be considered adequate to postpone surgical intervention, since historically these procedures have carried a high complication rate with subsequent penile deformities and impotence. The most conservative shunt is the Winter procedure or one of its modifications, whereby a needle or scalpel is inserted through the glans into one of the corpora and the viscous blood is aspirated. This temporarily allows for drainage of cavernous blood into the systemic circulation and has been successfully used in patients with SCA. Intermittent compression with a blood pressure cuff is critical to limit refilling. Anecdotal evidence suggests that penile implants to correct impotence may be more easily done soon after an impotence-causing episode.
Splenic Sequestration
Along with bacterial sepsis, historically one of the leading causes of death in children with SCA is the acute splenic sequestration crisis. Children with SCA who have not yet undergone autosplenectomy as well as older patients with other forms of SCD may develop sudden, rapid, massive enlargement of the spleen with trapping of a considerable portion of the red-cell mass. Patients suddenly become weak, pale, and dyspneic, with rapidly distending abdomen, left-sided abdominal pain, vomiting, and hypovolemic shock. The tempo of this crisis may be so fast that the patient dies before reaching the hospital. On physical examination there may be profound pallor plus hypotension with cardiac decompensation and massive tender splenomegaly. The Hb concentration is at least 2 g/dL lower than baseline, accompanied by a brisk reticulocytosis with increased nucleated red cells (distinguishing it from the aplastic crisis described below) and moderate to severe thrombocytopenia caused by splenic trapping of platelets. This complication has been described in patients as young as 8 weeks of age.
Emond described the natural history of acute sequestration crisis in a cohort of 308 children with SCA in Jamaica. Eighty-nine patients experienced 113 attacks, with 67 children having their first attack before the age of 2 years. There were 13 fatalities, 10 of which occurred before the age of 2. The most commonly associated clinical problem was upper respiratory tract infection and ACS. Sixteen percent of the patients had positive blood cultures. Recurrences were frequent, occurring in 49% of survivors of the first attack. A parental education program directed toward teaching parents the technique of spleen palpation and the urgency of seeking medical attention for enlarged spleen and pallor led to an increase in the incidence of cases (from 4.6 to 11.3 per 100 patient years of observation) and decreased the fatality rate (from 29.4 per 100 events to 3.1 per 100 events).
Therapy is the emergency restoration of intravascular volume and oxygen-carrying capacity by the immediate transfusion of packed red cells, even if only unmatched blood is available. Once normal cardiovascular status is restored, patients improve rapidly. The spleen usually shrinks within a few days, and the thrombocytopenia resolves. Care must be taken to not transfuse to baseline Hb, however, because with resolution the spleen releases the endogenous trapped erythrocytes and may result in a relative erythrocytosis with hyperviscosity, what is sometimes referred to as the “overshoot phenomenon” after simple transfusion. Recurrence of sequestration is common, usually within 2 to 3 months of the initial episode as the protection afforded by the transfused cells is lost. To eliminate recurrence some have recommended elective splenectomy after the first significant episode, whereas others more concerned with postsplenectomy sepsis have suggested splenectomy only after two episodes of sequestration. Emergent splenectomy during the acute sequestration is not indicated.
Kinney’s review from Duke University of splenic sequestration strengthens the position of those who favor elective splenectomy for children who have had one episode of acute sequestration, especially if the initial event was life-threatening. In this series of 23 patients, four underwent early splenectomy, seven were observed carefully, and 12 were placed on a maintenance transfusion program. Of those treated using transfusion, two were well and still receiving transfusion, three had recurrences while still on transfusion, four experienced recurrences within 3 months of the last transfusion, and only three remained well at 11 months (3.9 years after the last transfusion). Fourteen percent of children became alloimmunized, and 1 patient developed non-A, non-B hepatitis. Eventually 14 of the 23 patients underwent splenectomy. All were immunized against pneumococcus and were prescribed penicillin for prophylaxis. None had experienced a life-threatening infection 65.6 patient years later. Serjeant has demonstrated in a 22.5 year follow-up study that children in Jamaica who are postspenectomy are not at greater risks for infection than a matched control group of patients with SCA.
Rao has described a subacute form of splenic sequestration in 11 patients characterized by increased spleen size, 25% drop in hematocrit, less than 100,000 platelets, and reticulocyte count elevation above patient’s baseline. All responded to chronic transfusion programs, but seven patients had recurrent episodes after transfusions were stopped and eventually required splenectomy.
Aplastic Crisis
The clinical characteristics of the transient aplastic crisis of SCD result from the abrupt cessation of erythropoiesis, most commonly because of acute parvovirus infection. In the normal steady state, patients can compensate for the decreased red-cell survival (15 to 50 days) by increasing the bone marrow output six- to eightfold, with the absolute reticulocyte count sometimes reaching 400 to 500 × 10 9 /L. Temporary cessation of bone marrow activity because of suppression by intercurrent viral or bacterial infection causes the hematocrit value to fall as much as 10% to 15% per day with no compensatory reticulocytosis. The short-lived HbF-poor cells are the first to disappear from the circulation, whereas the cells containing more HbF persist. This natural selection of HbF-containing cells accounts for the apparent increase in the percentage of HbF during aplastic episodes.
Patients usually have pallor and fatigue but are without jaundice and have laboratory evidence of severe anemia below baseline with associated severe reticulocytopenia. Spontaneous recovery is usually heralded by a markedly elevated nucleated RBC count, followed in 1 or 2 days by a brisk reticulocytosis. Spontaneous recovery usually occurs within 7 to 10 days and may require no therapy, but some patients are extremely ill at presentation and many require short-term hospitalization and erythrocyte transfusions. This recovery phase, with the characteristic anemia, nucleated red cells, reticulocytosis, and occasional hyperbilirubinemia, may be responsible for most cases referred to as hyperhemolytic crisis.
In 1981, two groups linked transient aplastic crisis to parvovirus B19 infection. The Jamaican group subsequently documented the epidemiology and follow-up treatment of parvovirus in a cohort of infants identified at birth: 308 with SCA and 239 controls. They made a number of important observations: 1) the frequency of infection (about 40%) did not differ between sickle and control groups; 2) 20% of infections in the sickle group did not result in clinically recognizable aplasia; 3) 100% of aplastic episodes were associated with parvovirus; 4) no patients had recurrent aplasia; and 5) 45% of infected patients maintained an elevated parvovirus-specific IgG after 5 years. The secondary attack rate of siblings was 50% to 60%; therefore affected siblings should be monitored for evolving aplastic crisis when a child is diagnosed with an acute parvovirus B19 infection. Parvovirus B19 has also been implicated as the etiologic agent in erythema infectiosum (fifth disease), although often the classic rash of fifth disease is absent. Parvovirus B19 infection has also been related to several other sickle cell complications including acute splenic sequestration, hepatic sequestration, ACS, glomerulonephritis, and stroke. Many patients are seropositive for parvovirus B19 without history of a known aplastic crisis, suggesting many infections are subclinical. The relationship between parvovirus and erythropoiesis has been studied in detail. The virus specifically retards late erythroid precursor differentiation and is responsible for temporary erythroid aplasia in a broad array of hemolytic anemias.
Infection
Acute Infection
Bacterial infection is the most common cause of death in children with SCA. In one study the risk of acquiring sepsis or meningitis was higher than 15% in children younger than 5 years, with an associated mortality of approximately 30%. In young children the risk of pneumococcal sepsis appears to be 400 times that of normal children, and Haemophilus influenzae sepsis appears to be two to four times as common. These two reports are from the period before the licensing of conjugated vaccines to S. pneumoniae and H. influenzae. In general the organisms responsible for infection are primarily encapsulated bacteria and not unusual pathogens ( Table 20-2 ), but the infections that they cause in such patients occur more often and are more severe.
| Organism | Patient | Total No. Cases | Isolated Bacteremia | Acute Chest Syndrome | Meningitis | Bone/Joint |
|---|---|---|---|---|---|---|
| Streptococcus pneumoniae | SS | 62 | 39 (5 dead) | 14 (1 dead) | 8 (2 dead) | 1 |
| SC | 12 | 9 | 3 | 0 | 0 | |
| Haemophilus influenzae | SS | 10 | 6 (2 dead) | 3 | 1 | 0 |
| SC | 4 | 2 | 2 | 1 | 0 | |
| Staphylococcus aureus | SS | 5 | 2 | 2 | 0 | 1 |
| SC | 1 | 1 | 0 | 0 | 0 | |
| Viridans streptococci | SS | 5 | 4 | 1 | 0 | 0 |
| SC | 1 | 1 | 0 | 0 | 0 | |
| Escherichia coli | SS | 5 | 3 | 1 | 0 | 1 |
| SC | 2 | 0 | 1 | 0 | 1 | |
| Salmonella species | SS | 3 | 2 | 0 | 0 | 1 |
| SC | 2 | 1 | 0 | 0 | 1 | |
| Other | SS | 2 | 1 | 1 | 0 | 0 |
| SC | 0 | 0 | 0 | 0 | 0 |
The major risk factor for this increased vulnerability to infection is splenic dysfunction. The spleen normally serves two separate immunologic functions: 1) clearance of particles from the intravascular space, and 2) antibody synthesis. Both functions are impaired in SCD.
During the first year of life, “functional asplenia” develops in patients with SCD, particularly in those with SCA. This is heralded by the appearance of red cells with micronuclear remants (Howell-Jolly bodies) and intracellular vacuoles that appear as irregular surface characteristics (“pits”). When the percent of pitted red cells exceeds 3.5%, the spleen is generally considered nonfunctional. Splenic dysfunction develops early in life, with 50% of 2068 children with SCA having greater than 3.5% pitted red cells by 2 years of age. Appearance of dysfunction was less rapid and less common in children with HbSC or HbS β + -thalassemia. Despite palpable splenomegaly, children with elevated pitted RBCs have no splenic uptake of technetium-99m ( 99m Tc) sulfur colloid and are susceptible to the most serious infectious complication of asplenia and pneumococcal sepsis. Repeated infarction results in a nonpalpable, fibrotic, often calcified spleen. Splenic infarction in young children with SCA is an insidious process (usually with few symptoms) but is also progressive and culminates in splenic dysfunction. In older children and adults, especially those with HbSC and HbSβ + -thalassemia, severe left upper quadrant pain often accompanies acute splenic infarction. Transfusion, hydroxyurea, and bone-marrow transplantation can correct the splenic phagocytic defect, although these treatments are not indicated for splenic dysfunction per se.
Children with functional asplenia fail to respond to intravenously administered antigen vaccines, even when the phagocytic function of the spleen has been restored by transfusion. As with other individuals with asplenia, however, these children do respond normally to intramuscularly/subcutanously administered polysaccharide and conjugated pneumococcal vaccine.
Levels of serum immunoglobulins are normal or increased in children with SCA. However the serum is deficient in heat-labile opsonizing activity related to an abnormality of the properdin pathway, which is specific for the phagocytosis of pneumococci. There is an increased activation of complement via the alternate pathway but no intrinsic defect in the complement system. Opsonic activity can be reconstituted in vitro by addition of only the F(ab′) 2 fragments of capsular antibodies to S. pneumoniae. This abnormal opsonic activity and associated functional asplenia may partially explain the propensity for infections with pneumococcus and other encapsulated organisms.
Recurrent pnuemoccocal infections have been described in patients with SCD. In one series patients with serious invasive bacterial infections are significantly more likely to have recurrent infection.
Treatment of Infection
Because of the increased incidence of serious bacterial infections in patients with SCD, the index of suspicion for infection should always be high. In general the higher the temperature and leukocyte count, the higher the probability of a serious bacterial infection. Unfortunately however, the wide variability in the temperature and laboratory values of children with bacteremia does not permit an accurate prediction of whether an individual febrile child is bacteremic. The following guidelines are adapted from the NHLBI guidelines for empiric evaluation and treatment of the febrile child with a temperature higher than 101.3° F (38.5° C) :
- •
Perform complete blood cell count, reticulocyte count, blood culture. Consider urine and throat culture based on signs or symptoms. Perform chest radiography if pulmonary signs or symptoms are present.
- •
Prompt administration of broad-spectrum empiric parenteral antibiotics that provide coverage against S. pneumoniae and gram-negative enteric organisms before the results of laboratory tests are available or radiographs are taken. Empiric antibiotic selection should be based on the ability to kill both pneumococcus and H. influenzae and to penetrate the CNS. In areas where β-lactamase-producing H. influenzae or penicillin- and cephalosporin-resistant pneumococci are regularly encountered, these issues should be factored into the choice of antibiotic.
- •
Typically after antibiotic administration (e.g., ceftriaxone, 50 to 75 mg/kg, parenterally) the patient should be observed over 2 to 6 hours. Subsequent outpatient management is feasible in patients who do not appear to have toxic reactions or other features to consider for admission. Follow-up evaluation the next day and possibly repeat the empiric antibiotic dose. If children do well and culture results remain negative after 24 to 48 hours, antibiotics can be discontinued.
- •
Admission to hospital and continuation of empiric parenteral antibiotic treatment should be considered for:
- •
Appearance of toxicity
- •
Temperature higher than 40° C
- •
Infiltrate on chest radiograph
- •
WBC count higher than 30,000/mm 3 or below 500/mm 3 , platelet count less than 100,000/mm 3 , or Hb below 5 g/dL
- •
History of prior bacterial sepsis should be treated parenterally and patient should be admitted to the hospital.
- •
Previous surgical splenectomy
- •
Incomplete vaccination to pneumococcus or H. influenzae
- •
- •
Lumbar puncture should be performed on children having toxic reactions and those with any signs of meningitis. Bacterial meningitis should be treated for a minimum of 10 days parenterally or for at least 1 week after the cerebrospinal fluid has been sterilized.
- •
Documented sepsis should be treated parenterally for a minimum of 1 week.
- •
Patients with infiltrate on the chest radiograph should be treated as described for ACS. Because of the high incidence of atypical pathogens, patients should be treated with an appropriate antipneumococcal agent and macrolide antibiotic for at least a week.
Despite advance in laboratory and imaging techniques, the diagnosis of osteomyelitis in SCD is often difficult to distinguish from aseptic bone infarct and still relies heavily upon clinical assessment. Patients with clinical findings that are highly suggestive of osteomyelitis should have needle aspiration and culture of the lesion. After cultures have been obtained, children who appear acutely ill should be started on antibiotics. Antibiotic choice should include agents effective against Salmonella species and Staphylococcus aureus . Antibiotics should be discontinued or modified when culture reports are available.
Prevention of Infection
Methods of preventing invasive bacterial infection in SCA patients have focused on prophylactic antibiotics and vaccines. Prophylactic penicillin was first shown to be effective in the prevention of pneumococcal disease by John and associates, who used monthly intramuscular injections of long-acting penicillin. However if penicillin prophylaxis was discontinued after 3 years of age, an increase in pneumococcal infections was noted. Gaston and co-workers, in a blinded placebo-controlled clinical trial in the United States, showed that 84% of the pneumococcal infections in children younger than the age of 5 years could be prevented with oral penicillin. As a result of this landmark study, universal newborn screening has emerged throughout the United States, so that all children with SCA can be identified and started on penicillin prophylaxis by 3 months of age. If penicillin is discontinued after 5 years there is no increased incidence in infections compared to those who remained on prophylaxis; and there are conflicting data regarding the increased risks of penicillin-resistant pneumococcal colonization while on prophylaxis. Our approach to prophylaxis is as follows:
- •
Give prophylaxis to all newborns with HbSS, HbS β-thalassemia, and HbSC disease. (Some practitioners prefer not to treat infants with HbSC disease because their splenic dysfunction typically appears later in life.)
- •
Start as early as possible, optimally by 8 weeks of age but certainly by 3 months.
- •
Prescribe penicillin, 125 mg orally, twice a day until age 3 years. Then increase the dose to 250 mg orally, twice a day. Penicillin can be stopped after 5 years of age without resulting in an increased incidence of infection. Consider continuation for history of invasive bacteremia or surgical splenectomy.
- •
For patients allergic to penicillin, prescribe erythromycin, 10 mg/kg orally, twice a day.
- •
Educate families as to the importance of compliance and the early recognition of signs of infection, particularly temperature of 101.3° F (38.5° C) or higher.
- •
For vaccination recommendations, consult the Advisory Committee on Immunization Practices (ACIP) statements for detailed up-to-date recommendations for SCD. Complete routine vaccination schedule including complete conjugated vaccine series for S. pneumoniae (Prevnar-13; PCV13) and H. influenzae . Polyvalent pneumococcal vaccine has been shown to be effective in eliciting a normal antibody response, increases in pneumococcal opsonizing activity, and reducing the incidence of pneumococcal disease. The 23-valent polysaccharide pneumococcal vaccine (Pneumovax; PPV23) represents 90% of the common serotypes of pathogenic pneumococci, however, because children younger than 2 years have a poor response to this vaccine it should be given at age 2 years with a booster 3 to 5 years later. H. influenzae conjugated vaccine is immunogenic in children with SCD and should be given on the same schedule used for normal infants. Seasonal influenza vaccine should be administered to all SCD patients annually. Conjugated meningococcal vaccine (Menactra; MCV4) should be administered to patients at age 2 years.
Chronic Organ Damage
Cardiovascular System
Abnormal cardiac findings are present in most patients with SCA and are primarily the result of chronic anemia and the compensatory increased cardiac output. On physical examination, the most common finding is systolic ejection murmur, although concomitant S 3 , split S 1 , suprasternal notch thrill, and diastolic murmur may also be present. Echocardiography may be necessary to diagnose abnormal cardiac structure. Cardiomegaly is common, with electrocardiographic findings of left ventricular hypertrophy in about 50% of patients.
In view of the fact that the hallmark of SCD is vaso-occlusion, it is remarkable that myocardial infarction is an extremely rare event. In one review of the postmortem literature related to SCD including examination of 153 hearts, only four infarcts were reported. Despite the high oxygen extraction in the coronary circulation, blood in the coronary sinus contains no more sickled forms than the general circulation, presumably because of the short transit time through the coronary vessels. Atherosclerosis is virtually absent in this population, and the SCD heart has normal patent coronary arteries, often of larger caliber than seen in normal hearts.
The most comprehensive prospective examination of cardiac function in an unselected population of patients with SCA was done in the CSSCD. One hundred and ninety one patients ages 13 and older in steady state underwent echocardiography, and all of the measurements were performed centrally by an investigator without access to other patient data. After appropriately adjusting for body surface area, the left and right ventricles, aortic root, and left atrium were found to be larger than normal. Significant wall thickening was found only in the septum. The left ventricular dilation correlated with Hb and age, suggesting that the major cardiac findings are indeed related to the years of increased stroke volume in compensation for anemia and abnormal rheology.
PHT can occur in adult patients with SCD, both as a result of cardiac dysfunction and also from intrinsic pulmonary vasculopathy, with a reported prevalence as high as 20% to 40% based on tricuspid regurgitant jet velocity (TRJV) measurements. Multiple retrospective studies report a significantly increased mortality in patients with SCD and elevated TRJV compared with those patients without this finding. Castro et al. demonstrated that patients with SCD and PHT confirmed by right heart catheterization had median survival of 25.6 months, whereas patients without PHT had greater than 70% survival at the end of a 10-year observation period. A more recent study of 195 patients with SCD reported a significantly higher mortality rate for adult patients with TRJV (>2.5 m/s on echocardiography after a median follow-up length of 18 months). Increased lactate dehydroenase (LDH), bilirubin, and reticulocyte count have been associated with the elevated TRJV findings, which has led to a hypothesis that increased hemolysis is responsible for the vasculopathy. Because intravascular hemolysis leads to increased plasma Hb and arginase, which are associated reduced NO bioavailability (see Hemolysis and Nitric Oxide Depletion ), subsequent endothelial dysfunction and PHT could result from this pathophysiology. The prevalence of elevated TRJV in children appears similar to that in adults ; however, a recent French adult study revealed a prevalence of 27%, but confirmation by catheterization revealed that PHT prevalence was only 6%. Therefore the positive predictive value of TRJV greater than 2.5 m/s appears low. Further concerns about the reproducibility of TRJV measurements in children, as well as the prognostic implications that differ from adults, have prevented the development of screening recommendations among children with SCA.
The steady-state blood pressure in patients with SCA (and to a lesser extent all forms of SCD) is lower than in a race-, gender-, and age-matched normal population ( Table 20-3 ). This observation may reflect differences in peripheral vascular resistance or be related to the tendency to lose sodium and water in the urine. Higher blood pressure is a risk factor for stroke, early mortality, and nephropathy, but the diagnosis of elevated blood pressure depends on knowing the normal values for this population (see Table 20-3 ). The clinician should note that blood pressures that appear to be normal based on race, gender, and age could be significantly elevated and clinically ominous in the sickle population. Such patients should be considered candidates for antihypertensive therapy.
| Percentile | Age (yr) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-3 | 4-5 | 6-7 | 8-9 | 10-11 | 12-13 | 14-15 | 16-17 | 18-24 | 25-34 | 35-44 | ||
| Females | ||||||||||||
| Systolic | 50 | 90 | 95 | 96 | 96 | 104 | 106 | 110 | 110 | 110 | 110 | 110 |
| 90 | 100 | 110 | 110 | 110 | 110 | 118 | 120 | 122 | 122 | 125 | 130 | |
| Diastolic | 50 | 52 | 60 | 60 | 60 | 60 | 62 | 70 | 70 | 64 | 68 | 70 |
| 90 | 62 | 70 | 70 | 70 | 74 | 74 | 80 | 78 | 80 | 80 | 84 | |
| Males | ||||||||||||
| Systolic | 50 | 90 | 95 | 100 | 100 | 100 | 110 | 108 | 112 | 112 | 114 | 110 |
| 90 | 104 | 110 | 108 | 116 | 112 | 120 | 120 | 128 | 130 | 130 | 132 | |
| Diastolic | 50 | 54 | 60 | 60 | 60 | 60 | 64 | 64 | 70 | 68 | 70 | 70 |
| 90 | 66 | 68 | 68 | 70 | 70 | 72 | 78 | 80 | 80 | 80 | 84 | |
Renal System
Hyposthenuria, hematuria, nephrotic syndrome, and uremia are the major renal complications of SCD. In addition the production of erythropoietin in response to anemia may be lower in older patients with SCA, possibly because of primary renal disease.
Hyposthenuria develops early in childhood, and as is the case with functional asplenia, may be temporarily reversed with transfusion. The hypertonic environment of the renal medulla promotes intraparenchymal sickling even at normal pO 2 , which leads to decreased medullary blood flow and derangement of the countercurrent multiplier. Abnormality of the countercurrent multiplier may be the mechanism for hyposthenuria, or as suggested by Buckalew and Someren, it may be the result of decreased flow to nephrons with long loops of Henle and preservation of flow to nephrons with short loops. The obligatory water loss in SCA results in a tendency toward dehydration and invalidates the use of urine volume or specific gravity as an indicator of the patient’s state of hydration. Nocturia and enuresis are common complaints of these patients, who excrete large volumes of dilute urine. Urinary sodium losses may be high and result in significant hyponatremia. A renal tubular acidification defect as well as hyporeninemic hypoaldosteronism and impaired potassium excretion have been identified. In one review deJong and Van Eps emphasized that renal vasodilating prostaglandins are increased in patients with SCD, leading to a compensatory increase in renal blood flow, glomerular filtration rate (GFR), and proximal tubular activity.
Although hematuria is usually mild, overt bleeding is occasionally severe enough to cause significant blood loss. Papillary necrosis is usually the underlying anatomic defect. Maintenance of high urinary flow with increased fluid intake will eliminate clots from the bladder and decrease medullary osmolality. Urinary alkalinization and diuretics may be beneficial until the bleeding stops. ε-Aminocaproic acid has been suggested to treat severe hematuria that is refractory to supportive care and transfusion, but it must be used cautiously because of the risk of ureteral or pelvic clotting and obstruction. In patients with long-standing hematuria, supplemental iron may be necessary to prevent iron deficiency. Hematuria may also be the presenting symptom of a renal tumor. A surprisingly high incidence of renal medulary carcinoma has been reported in patients with SCD and even persons with sickle trait.
Uremia is a rare complication in children with SCD that may follow a symptom complex of nephrotic syndrome with glomerulonephritis. The nature of the glomerular lesion is unknown and may represent response to iron deposition, antigen-antibody complex, or mesangial phagocytosis of fragmented sickled cells. Recently an association between parvovirus infection with or without an antecedent aplastic crises has been associated with acute glomerulonephritis.
Proteinuria in SCD begins in childhood and teenage years and can progress to nephrotic syndrome and ultimately to renal failure, which is a common cause of mortality among adults with SCA. Microalbuminuria manifested by a ratio of albumin to creatinine (>20 mg/g Cr) is common; a study of 442 children with SCD studied for 10 years reported proteinuria in 6.2% of children and 12% of teenagers. Proteinuria was associated with lower Hb levels, higher MCV, higher white cell counts, and more clinically severe disease. A study of 381 adults with SCD demonstrated that 7% had elevated serum creatinine levels and 26% had proteinuria. Ten patients with proteinuria underwent renal biopsy, and the glomerular lesions showed perihilar focal segmental sclerosis and glomerular enlargement, similar to findings in an animal model with glomerular hypertension and efferent arteriolar vasoconstriction. To test the animal analogy, these patients were treated with enalapril, an angiotensin-converting enzyme (ACE) inhibitor that has been shown to decrease efferent arteriolar constriction. In all treated patients the level of proteinuria fell during treatment and returned toward abnormal after discontinuation.
The predictive value of microalbuminria screening has not been fully elucidated in children but has been suggested by some. Serum cystatin C may be a better screen for GFR than creatinine in SCA, because it appears to rise according to the degree of albuminuria and before significant elevation in serum creatinine is detected.
Treatment of microalbuminuria or proteinuria with ACE inhibitors can be effective, but long-term therapeutic benefits for the prevention of progressive glomerular disease have not yet been demonstrated. Addition of hydroxyurea to ACE inhibitors may further improve renal function.
Bakir and associates estimate that 4% of adult patients with SCA develop nephrosis and that two thirds of these patients go on to develop renal failure. Renal failure can be managed with peritoneal dialysis, hemodialysis, and transplantation. Renal transplantation can be successful in patients with end-stage sickle nephropathy, but three-year allograft and patient survival are significantly decreased compared with other patient populations.
Hepatobiliary System
Liver and biliary tract abnormalities are common in SCD and are the result of cholelithiasis, hepatic infarction, and transfusion-related hepatitis and hemosiderosis.
Bilirubin gallstones are common, and two large series have studied the incidence of gallstones as detected by ultrasound in children with SCA. The percentage of gallstones in 226 patients aged 5 to 13 years selected randomly from a group of children with HbSS disease identified at birth was 13%, a value lower than that found in a survey of clinic patients studied by Sarnaik and associates. In this report, the incidence of gallstones was 12% by 2 to 4 years of age. With advancing age, the incidence increased gradually, reaching 42% in the 15- to 18-year-old age group. Fourteen of the 226 patients were noted to have “sludge” in the gallbladder, and on repeated ultrasonograms up to 2 years later, four had developed stones, 4 had no further evidence of sludge, and 6 remained unchanged. Comorbid Gilbert syndrome (uridine diphosphate [UDP]–glucuronosyltransferase 1A1 [UGT1A1] promoter polymorphism) significantly increases the serum bilirubin levels in children with SCD and the likelihood of requiring early cholecystectomy. Evidence is good that children tolerate elective cholecystectomy with little morbidity if they are prepared properly for surgery. Laparoscopic cholecystectomy has been particularly effective in reducing the postoperative hospital stay in children with SCD. In contrast operating during the acute phase carries a significant risk of complication.
Intrahepatic sickling can result in massive hyperbilirubinemia, elevated liver enzyme values, and a painful syndrome mimicking acute cholecystitis or viral hepatitis. (See “ Acute Abdominal Pain ”). Fulminant hepatic failure with massive cholestasis and rapidly progressing hepatic encephalopathy and shock has been described as a rare but often fatal complication of SCD that may be amenable to exchange transfusion.
Eyes
SCD can affect virtually every vascular bed in the eye. Tortuosity and sacculation of conjunctival vessels are seen in more than 90% of patients with SCD. These lesions are best seen in the lower temporal area, disappear after exchange transfusion, and are curiously related to the ISC count in the peripheral blood. They are purely cosmetic, however, and have no deleterious effect on the eye.
Sickle retinopathy is classified as either proliferative or nonproliferative. Nonproliferative retinopathy probably results from retinal arteriolar infarction with adjacent hemorrhage and requires no therapy. Depending on the age, layer, and extension of the hemorrhage, the result can be a salmon patch, schisis cavity, vitreous hemorrhage, or black sunburst. In two patients with documented acute arteriolar occlusion, salmon patches developed in a matter of hours to days, with atrophic schisms cavities evolving in 3 to 4 months. In older patients angioid streaks are common, but the cause is unknown.
The more serious complication is proliferative retinopathy, which has been classified by Goldberg as stage 1, peripheral arteriolar occlusions; stage 2, arteriolar-venular anastomoses; stage 3, neovascularization; stage 4, vitreous hemorrhages; and stage 5, retinal detachment. Because these lesions may progress to blindness, laser therapy to occlude feeding vessels of advanced proliferative lesions has been advocated. Unfortunately photocoagulation carries the risks of neovascularization of the choroid and retinal breaks, and complications that can result in blindness. The dilemma of choosing potentially blinding therapy for a potentially blinding lesion is complicated by the observation that some proliferative lesions heal spontaneously by autoinfarction. In one study of untreated sickle retinopathy in Jamaica, 567 eyes were observed for 8 years. Proliferative retinopathy was initially present in 12% of the eyes: another 8% developed retinopathy during the follow-up study, and patients with HbSC were more commonly affected than those with SCA. Blindness resulted in 12% of the eyes with retinopathy. In the original group of eyes with retinopathy, 30% developed progressive retinopathy, 10% showed spontaneous regression, and the remaining 30% showed a mix of regression and progression. In another prospective Jamaican study, treatment of retinopathy was compared with no treatment. No statistical difference in visual acuity between the two groups was reported. Macular ischemia and color blindness have been reported to be prevalent in patients with SCD without evidence on ophthalmologic examination of retinal lesions.
Rarely acute painless loss of vision is the result of central retinal artery occlusion. Although such lesions may resolve spontaneously, exchange transfusion has been recommended for bilateral disease.
Blunt trauma to the eye may result in hyphema (bleeding into the anterior chamber). Because the conditions in this chamber overwhelmingly favor sickling, any HbS-containing red cell will sickle (even sickle trait) and may cause obstructive glaucoma and blindness. Common medical treatments that might be considered in patients without SCD may promote sickling and should be avoided (e.g., hyperosmotic/diuretic agents/carbonic anhydrase inhibitors). Initial topical β-adrenergic antagonists (e.g., timolol) or α 2 -adrenergic agonists (e.g., brimonidine) may be effective, but a low threshold for surgical evacuation of anterior chamber blood should be observed so that vision is preserved. This condition is one of the true ocular emergencies that occurs in patients with SCD as well as sickle trait.
Skin
Leg ulcers usually do not occur in childhood, but in adolescence and adulthood ulcers may constitute a crippling symptom. This skin lesion is also very rarely seen in other chronic hemolytic anemias such as hereditary spherocytosis, thalassemia, elliptocytosis, and pyruvate kinase deficiency and therefore may not represent a vaso-occlusive phenomenon. Ulceration may result from increased venous pressure in the legs caused by the expanded blood volume in the hypertrophied bone marrow. In tropical areas in which shoes are not usually worn and insect bites are common, leg ulcers are also common. In Jamaica, leg ulcers typically start in the 10- to 20-year-old group and eventually appear in 75% of adults. Koshy and colleagues have reported data from the CSSCD regarding leg ulcers in patients with SCD. Leg ulcers appear to be less common in individuals with two α-globin genes than with those with three or four genes. The incidence of leg ulcers appears to decrease consistently with increases in HbF production. There is decreased venous outflow in in the lower extremities of patients with leg ulcers compared with patients without ulcers, suggesting that venous incompetence may contribute to the development or failure to heal of these lesions. In addition, low steady-state Hb values and markers of high hemolytic rate are associated with increased incidence of leg ulcers and may be associated with decreased NO bioavailability and endothelial dysfunction.
Chronic leg ulcers become a major source of morbidity and have a profound negative impact on educational achievement and employment. Usually present over the medial surface of the lower tibia or just posterior to the medial malleolus, they begin as a small depression with central necrosis and, if unattended, widen to encircle the entire lower leg. Debridement, scrupulous hygiene, topical antibiotics, bed rest, and elevation of the leg are the mainstays of therapy. In some patients, protection of the ulcer by the application of a soft sponge-rubber doughnut and a low-pressure elastic bandage seems to be beneficial. One report suggested that an arginine-glycine-aspartic acid (RGD) peptide matrix designed to mimic the normal matrix was beneficial. Close attention to improved venous circulation by the use of above-the-knee elastic stockings may prevent ulceration. If ulcers persist despite optimal care, transfusion therapy may be used and consideration given to split-thickness skin grafts. Transfusion therapy is sometimes effective, but in many patients the ulcers either do not heal or recur after discontinuation of this therapy. Oral zinc sulfate may promote healing of leg ulcers, and peripheral vasodilator therapy appears ineffective.
Skeleton
Skeletal changes in SCD are common and are the result of expansion of the marrow cavity and repeated bone infarction. The expanded marrow is best seen in radiographs of the thickened calvarium with a wide diploic space. Overgrowth of the anterior maxilla may lead to severe orthodontic and cosmetic problems. Vertebrae are generally flattened, with a characteristic biconcave deformity called codfish vertebrae. In older patients, vertebral disease may cause chronic back pain. These individuals need to be treated as other patients who have chronic back disease with appropriate exercises, braces, bed rest, muscle relaxants, and moral support.
The major chronic bone complication of SCD is AVN. Indeed, the most common etiology of AVN of the femoral head in children is SCD. As shown ( Fig. 20-8 ), the incidence is relatively low in children, with a prevalence estimated at 3% in children under 15 years, but increases with age and occurs in both patients with SCA and HbSC disease. The incidence is remarkably increased in patients with SCA and α-thalassemia, who also have higher hematocrit levels and rates of pain. Athophysiology of AVN is unknown but hypothesized to result from limited blood supply in the femoral (and humeral) heads that promotes sludging in marrow sinusoids, marrow necrosis, healing with increased intramedullary pressure, articular surface necrosis, bone resorption, and eventually epiphyseal collapse. The diagnosis is often made clinically with pain and limited range of motion or radiographically, classically with a spectrum of findings from subepiphyseal lucency and widened joint space to flattening or fragmentation and scarring of the epiphysis. Although roughly half of the patients diagnosed on the basis of screening radiographs were clinically asymptomatic, significant chronic pain and limited joint mobility plagued the others. MRI can identify AVN changes earlier than conventional films.