In Africa, at least 240,000 children are born each year with sickle cell disease. Historically, in the absence of newborn screening and appropriate treatment, most such children died undiagnosed in early childhood. However, with increasing awareness of the condition and economic and epidemiologic transition, increasing numbers are surviving. Greater investments in basic and applied research in the African context, and increased sensitization or African ministries of health regarding the importance of this condition, could make a substantial difference to the lives and livelihoods of millions of people living with sickle cell disease on the continent and their families.
Key points
- •
Sickle cell disease is a common and growing health problem in many parts of sub-Saharan Africa (SSA), where at least 240,000 affected children are born with the condition every year.
- •
Sickle cell disease is widely neglected on the continent, where an estimated 50% to 90% of those born with the condition die undiagnosed before their fifth birthdays.
- •
An unknown, but probably large, proportion of these deaths are almost certainly attributable to 2 main conditions: malaria and invasive bacterial infections.
- •
With economic and public health advancements in many parts of the region, survival of affected children is likely to improve, which will lead to a growing need for appropriate medical services.
- •
A greater emphasis on basic and applied research in the area of sickle cell disease in SSA could lead to substantial improvements to the lives and livelihoods of millions of affected people and their families.
Introduction
Sickle hemoglobin (HbS) is a structural variant of normal adult hemoglobin (HbA; α 2 β 2 ) in which the normal beta-globin subunit is replaced by a mutant form of the molecule (β S ) in which the glutamic acid residue normally present at position 6 is replaced by a valine residue, the result of a single nucleotide polymorphism (thymine to adenine; rs334) at position 17 of the HBB gene. This abnormal HbS polymerizes reversibly under low oxygen tension, and alterations to the shape, rheological properties, and membrane properties of the red blood cells that result from these polymerization events are central to the pathophysiology of the resultant disease. The term sickle cell disease (SCD) refers to a heterogeneous group of conditions in which HbS predominates. The most common form of SCD results from the homozygous inheritance of the β S mutation, a condition most commonly referred to as either HbSS or sickle cell anemia (SCA). Although SCA is responsible for at least 70% of SCD globally, SCD can also result from compound heterozygosity for HbS in association with a wide range of other HBB mutations, including the mutation that results in the production of another structural variant, hemoglobin β C (HbSC) and one of the many β-thalassemia mutations that lead to the reduced production of normal beta-globin (HbS/β-thalassemia). Although the geographic range of HbS extends throughout most of sub-Saharan Africa (SSA) north of the Zambezi river, both HbC and β-thalassemia are confined to more limited parts of West Africa and to the historic trade routes of North Africa. As a result, HbSS is by far the most significant form of SCD in SSA, and the form of SCD about which most is known. HbSS is therefore the main focus of this article.
Introduction
Sickle hemoglobin (HbS) is a structural variant of normal adult hemoglobin (HbA; α 2 β 2 ) in which the normal beta-globin subunit is replaced by a mutant form of the molecule (β S ) in which the glutamic acid residue normally present at position 6 is replaced by a valine residue, the result of a single nucleotide polymorphism (thymine to adenine; rs334) at position 17 of the HBB gene. This abnormal HbS polymerizes reversibly under low oxygen tension, and alterations to the shape, rheological properties, and membrane properties of the red blood cells that result from these polymerization events are central to the pathophysiology of the resultant disease. The term sickle cell disease (SCD) refers to a heterogeneous group of conditions in which HbS predominates. The most common form of SCD results from the homozygous inheritance of the β S mutation, a condition most commonly referred to as either HbSS or sickle cell anemia (SCA). Although SCA is responsible for at least 70% of SCD globally, SCD can also result from compound heterozygosity for HbS in association with a wide range of other HBB mutations, including the mutation that results in the production of another structural variant, hemoglobin β C (HbSC) and one of the many β-thalassemia mutations that lead to the reduced production of normal beta-globin (HbS/β-thalassemia). Although the geographic range of HbS extends throughout most of sub-Saharan Africa (SSA) north of the Zambezi river, both HbC and β-thalassemia are confined to more limited parts of West Africa and to the historic trade routes of North Africa. As a result, HbSS is by far the most significant form of SCD in SSA, and the form of SCD about which most is known. HbSS is therefore the main focus of this article.
Origins of the sickle mutation
Haplotype analysis suggests that the rs334 allele that encodes for β s has arisen, and been independently amplified to its current population frequencies, on at least 2 and likely more occasions. Despite being detrimental in its homozygous form (HbSS) the rs334 allele has reached high population frequencies throughout much of SSA to the extent that through much of the continent more than 15% of the population are heterozygotes (HbAS; sickle cell trait), and notably more than twice that in small surveys from selected populations. That such high heterozygote frequencies might result from selection for HbAS through a survival advantage against malaria was first suggested more than 6 decades ago and, despite some early skepticism, this hypothesis has since been confirmed beyond any reasonable doubt (reviewed in Ref. ). In a recent meta-analysis of available data from 44 studies conducted throughout the continent, Taylor and colleagues estimated that children with HbAS are more than 90% less likely to develop severe and complicated Plasmodium falciparum malaria, the form of malaria associated with the most deaths, than normal children with HbAA. This conclusion has recently been reaffirmed in the most substantial study of its kind conducted to date, involving almost 12,000 children with severe malaria and more than 17,000 controls recruited from 12 sites throughout the malaria-endemic world, in which the odds ratio (OR) for severe malaria among HbAS children was 0.14 and was associated with a significance level rarely seen in such studies ( P = 1.6 × 10 −225 ). Moreover, the effect of HbAS is not limited to the most severe forms of malaria but also extends to protection against uncomplicated forms of the disease, with the result that HbAS confers even wider health benefits and survival advantages by protecting against the longer-term consequences of uncomplicated malaria, such as chronic anemia, malnutrition, and invasive bacterial infections.
The precise mechanism by which HbS protects against malaria remains a subject of some speculation. Early work suggested that erythrocytes containing HbS might be less supportive of P falciparum growth and multiplication than normal red cells under low oxygen tension, but more recently it has been suggested that HbS might protect against malaria by mediating the reduced display of the parasite-encoded protein P falciparum erythrocyte membrane protein-1 (PfEMP1) on the surface of malaria-infected erythrocytes. The adherence of P falciparum –infected red blood cells to capillary endothelium has been implicated in both the pathogenesis of severe malaria and in the evasion of parasite-infected red blood cells from immunologic removal by the spleen, and so it has been speculated that reduced PfEMP1 display might result in both fewer pathologic consequences and improved clearance of infected red cells during malaria infections. In addition, it has also been suggested that parasite-infected HbS-containing erythrocytes may be removed more rapidly from circulation through innate or acquired immune-mediated processes ; a process that might also result in their premature destruction by the spleen.
Sickle cell disease in sub-Saharan Africa
The corollary of positive selection for HbAS through a survival advantage against malaria is that a small but variable proportion of children in malaria-endemic parts of SSA are born with either HbSS or with compound heterozygosity for both HbS and a second mutation that, together with HbS, results in SCD, the most notable being HbSC and HbS/β-thalassemia. Historically, SCD has nevertheless been profoundly neglected in the African context and even today the knowledge of a range of basic facts regarding the condition within the region is decades behind that for regions where SCD is considerably less common.
Despite the publication of a growing number of reports on the prevalence and distribution of the sickle cell trait in Africa toward the middle of the last century, SCD was rarely recognized by physicians on the continent even 40 years after the condition was first described in the United States. This remarkable observation even led some clinicians to question whether the mode of inheritance of SCD might be different in the African context. Even now, in the absence of newborn screening across much of the continent, there are no accurate, up-to-date figures regarding the survival rates for SCD in SSA, but, by analyzing population data on the age-specific prevalence of SCA, an indirect measure of the loss through death of patients with this condition, it was recently concluded that mortality among children less than 5 years old born with SCA in SSA remains unacceptably high at between 50% and 90%. These figures are in stark contrast to those from the north, where in recent years many countries have adopted universal screening for SCD and where most now provide comprehensive care for affected individuals. As a result, mortality is now rare among children born with SCD in Europe, the United States, and the Caribbean, where most affected children live to their 40s and 50s.
Because official statistics are so poor, even basic facts such as the number of births of children with SCD in Africa can only be estimated using indirect approaches. For example, we recently used a geostatistical model that combined data on the population frequencies of HbAS from the published and unpublished literature, and birth rates and population densities based on United Nations figures, to estimate birth rates for SCA (which accounts for approximately 70% of SCD) for every country in the world. We concluded that 312,000 (294,000−330,000) children were born with SCA globally in 2010, that almost 80% of these children were born in SSA, and that half were born in just 3 countries: Nigeria, the Democratic Republic of Congo, and India ( Fig. 1 ). Nevertheless, the veracity of these estimates is undermined by the paucity of data on which they are based, and given the high rates of consanguinity in many parts of the region the true numbers could be substantially larger. In combination, the high number of affected births and the high rate of early mortality mean that SCD is probably responsible for more than 6% of all child deaths in many parts of SSA.

The natural history of sickle cell disease in Africa
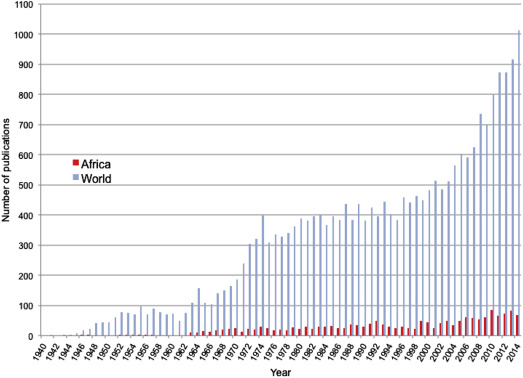
When the very high birth and death rates for children with SCD in Africa are considered, it is notable how poorly the natural history of the condition has been documented on the continent. Despite almost 80% of SCD births occurring in SSA (see Fig. 1 ), academic research from the continent lags far behind that from the north. Less than 7% of all articles published on the subject since 1940 have included the word Africa, and, even now, research output on SCD from Africa is at a similar level to that of the north from more than 60 years ago ( Fig. 2 ). Until recently, there were no programs for newborn screening, and no comprehensive studies of the natural history of SCD in Africa have been undertaken. As a result, the common causes of death among children with SCD on the continent have remained a matter of speculation, although it is likely that a substantial proportion of deaths have been attributable to just 2 conditions: malaria and invasive bacterial infections.
Malaria
Although it has frequently been assumed that malaria is a common cause for death in African patients with SCD, few reliable data are available on which to base this conclusion.
In light of the protective effects of HbS against malaria, which seems to be directly proportional to the intracellular concentrations of HbS in heterozygotes, it might be expected that homozygotes would be even more protected. However, there are also reasons why malaria might cause more problems in patients with SCD than in normal people. For example, the marked reticulocytosis seen in such patients might increase the efficiency of infection by P falciparum parasites, which show a preference for the youngest, most metabolically active red blood cells. Moreover, the characteristic hyposplenia seen in patients with SCA could lead to reduced clearance of infected red blood cells. With steady state hemoglobin concentrations that are typically in the rage of 6 to 8 g/dL, patients with SCA might also be vulnerable to the sudden onset of catastrophic anemia brought on by the hemolysis or ineffective erythropoiesis that is commonly associated with malaria infections, and it seems likely that the tissue hypoxia and inflammatory processes that typically accompany malaria infection might be potent triggers of sequestration crises. Nevertheless, clinical data relating to the relative risk of malaria and its consequences in patients with SCD are confusing.
Although malaria did not feature strongly in early descriptions of the natural history of SCD, it was implicated as a significant problem in several later reports. Nevertheless, although such reports confirmed that children with SCD can have malaria infections, they shed no light on the relative risks or consequences of the disease compared with normal children. Moreover, given the perception that malaria can be dangerous in patients with SCD, malaria prophylaxis is prescribed in most SCD clinics in malaria-endemic areas, making data from prospective cohort studies difficult to interpret. Through a birth cohort study conducted in western Kenya, in which participants were tested retrospectively for SCD on completion of the study (reducing the bias of prophylactic treatment) it was shown that, although all-cause mortality was significantly higher among children with SCD than control children, the incidence rates of both severe malaria anemia and high-density P falciparum infections were lower in children with SCD than in normal children. This finding suggests that children with SCD might have a degree of resistance to P falciparum infections but does not exclude the possibility that malaria infection might have been a precipitating event in some or all of those who died.
Intervention studies potentially offer a more robust approach to investigating the importance of malaria as a health problem in patients with SCD. Several placebo-controlled trials of antimalarial prophylaxis have been conducted in patients with SCD, including 2 considered sufficiently robust to be included in a recent Cochrane Review. The investigators concluded that malaria prophylaxis reduces the risks of sickle cell crises, blood transfusions, and hospital admissions, and results in increased mean hemoglobin levels, all of which suggest that malaria is an important cause of ill health among children with SCD in many parts of Africa and that such children should be protected from the disease.
Although the literature on the relationship between malaria and SCD remains confusing, the following describes the most likely situation. SCD is associated with protection against the acquisition of P falciparum infections, to a degree that may even be higher than the protection that is associated with HbAS. However, this protection is not complete, and when patients with SCD do become infected by malaria parasites the consequences can be catastrophic, through the precipitation of crises and the rapid development of severe anemia. With this in mind, it seems highly likely that, by diagnosing SCD through screening in early life and protecting affected children from malaria, the survival of children living with SCD in areas of high malaria transmission will be significantly improved. However, important questions still remain about the most appropriate strategies for malaria prevention. For example, should all malaria-exposed patients be prescribed prophylaxis throughout life, or is it possible that, at lower levels of malaria transmission, the risks of treatment might outweigh the risks of infection? With increasing resistance to antimalarial drugs and side effects from the long-term use of some, which antimalarial drugs offer the best balance between risk and benefit? Might alternative approaches, such as intermittent presumptive treatment of malaria or transmission avoidance through methods such as impregnated bed nets or indoor residual spraying of houses, be more appropriate in areas of lower transmission? Definitive clinical trials are needed to address these important questions.
Bacteremia
Invasive bacterial infections are a second group of conditions that make a significant but unknown contribution to the high early mortality seen in African children with SCD. Children with SCD manifest several immunologic abnormalities, including decreased splenic function, reduced serum opsonin activity, and abnormal neutrophil function, and it has long been known that children with SCA in the north are at considerable risk of invasive bacterial infections, particularly those caused by capsulated organisms such as Streptococcus pneumoniae , Haemophilus influenzae , and non-Typhi Salmonella species. Early studies conducted in the north showed that the risk of invasive pneumococcal disease during early life was up to 50-fold higher in children with SCD compared with normal children, and the subsequent implementation of strategies such as pneumococcal vaccination and penicillin prophylaxis led to the virtual elimination of this excess risk in multiple populations in subsequent years.
Despite being well documented in the north, data regarding the risks of bacteremia among patients with SCD in Africa have been slow to accumulate and subject to controversy. S pneumoniae was the most common organism isolated from children with SCD in an early study conducted in the Congo but subsequent studies conducted in Uganda and Nigeria suggested that other organisms, particularly Staphylococcus aureus , Escherichia coli , Klebsiella spp, and non-Typhi Salmonella , may be more important in the African context. In these studies, both S pneumoniae and H influenzae were found infrequently, leading some commentators to question the potential benefits of antimicrobial prophylaxis in African patients with SCD. Nevertheless, most of these studies were open to misinterpretation, because serious bacterial infections have their peak incidence in very early childhood, whereas most of the African studies were conducted in older children who survived their early years to present with symptoms of SCD. Furthermore, organisms like S pneumoniae and H influenzae are considerably more fastidious and difficult to grow than some of these other organisms, and culture sensitivity may therefore have biased the distribution of the pathogens observed in some of these studies.
Despite this early confusion, it has now been shown beyond all doubt that, just like children with SCD born in the north, children born with SCD in Africa are at substantial risk of severe infections in early life and that the highest risks relate to the same pathogenic organisms: S pneumoniae , H influenzae , and non-Typhi Salmonella species. Taking as a starting point more than 1700 children who were diagnosed with bacteremia through the surveillance of all admissions to a general pediatric facility, it was shown through retrospective genotyping for SCD that young children with the condition were at very high risk of bacteremia from these top 3 organisms (1.2–5.0 episodes/100 person years), a risk that was similar to that reported from developed countries (1.5–11.6 episodes/100 person years) during the era before the introduction of antibiotic prophylaxis or pneumococcal vaccines. A subsequent meta-analysis of available case-control studies conducted in African children showed that the risk of all-cause invasive bacterial infections was 19 times higher in children with SCD compared with normal children, 36 times higher for S pneumoniae , and 13-times higher for H influenzae type b. Collectively these observations suggest that invasive bacterial diseases make a significant contribution to the high mortality associated with SCA in African children.
The future of sickle cell disease in sub-Saharan Africa
Despite the historically low priority that SCD has had throughout much of SSA, there are promising signs that the situation is now beginning to improve. SCD has recently been recognized as an important but neglected problem by several key agencies, including the United Nations and the World Health Organization, and a growing number of research groups and governments are taking an increasingly active interest in the detection and management of the condition (see Fig. 2 ). Moreover, in recent years, pilot projects of newborn screening have been reported from several centers, which suggest that, in principle, early detection is within the reach of many countries in the region. Because the highest rates of mortality are likely to occur in the first few years of life, establishing such services in areas with the highest prevalence of SCD will be key to reducing the current levels of mortality in affected patients, and in many parts of Africa might also have a substantial effect on child mortality overall. The gradual introduction of newborn screening for SCD in the United States and many parts of Europe in the last 40 years, coupled with the provision of a basic package of care (including vaccination for common bacterial infections and penicillin prophylaxis until at least 5 years of age), has led to a situation in which most children born with SCD in the north can now expect to live until their 40s and 50s. However, as discussed later, to assume that similar advances could be achieved in most parts of Africa without substantial education programs, the careful fostering of political will, and significant financial investment, would be simplistic.
Related posts:
 The Global Burden of Anemia
The Global Burden of Anemia
 Problems and Approaches for Blood Transfusion in the Developing Countries
Problems and Approaches for Blood Transfusion in the Developing Countries
 Hematological Practice in India
Hematological Practice in India
 Glucose-6-Phosphate Dehydrogenase Deficiency
Global Approach to Hematologic Malignancies
Improving Laboratory and Clinical Hematology Services in Resource Limited Settings
Glucose-6-Phosphate Dehydrogenase Deficiency
Global Approach to Hematologic Malignancies
Improving Laboratory and Clinical Hematology Services in Resource Limited Settings
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


