Fig. 3.1
Generation and inflammatory cellular targets of extracellular heme in sickle cell disease. Hemolysis releases cell-free hemoglobin (Hb), which is normally scavenged by haptoglobin and CD163. Free hemoglobin reacts with and scavenges NO via the dioxygenation reaction and also reacts with hydrogen peroxide to generate hydroxyl radicals via the Fenton reaction. Oxidized hemoglobin releases free heme, which can activate the endothelium to increases adhesion molecule expression, and stimulates neutrophils to release NETs
The role and mechanism of heme in the pathobiology of SCD is complex. Intra-peritoneal injections of purified heme exert a salutary effect on vascular stasis in sickle mice by inducing HO-1 [79]. Meanwhile, intravenous injection elevates the extracellular heme pool, depletes plasma hemopexin, and induces a lethal ALI [80]. The latter effect is typical of DAMP molecules—intracellular molecules that elicit robust inflammatory responses when they are released externally by tissue damage [81, 82]. Although heme was already known to be recognized by toll-like receptor 4 (TLR4) [83], elucidation of its ALI inducing effect has helped to define its role as a bona fide DAMP molecule. It is arguably the prototypical erythroid DAMP. Intravenous infusion of a modest dose of purified heme caused sudden death in SS mice; autopsies revealed erythrocyte congestion in the lungs suggesting that VOC in the pulmonary microvasculature contributed to this lethality. A model in which the extracellular heme/TLR4 signaling axis activates NF-kappa B phospho p65 to cause VOC through increased adhesion molecule expression and degranulation of Weibel Palade bodies has been proposed [58]. The reverse sequence (i.e. VOC → hemolysis) is well established in humans with SCD, even among those in whom the acute VOC does not progress to a life threatening condition such as ACS [66]. Thus, it appears that VOC and intravascular hemolysis are intertwined in a vicious cycle that drives much of the pathobiology of SCD.
Extracellular Heme Crisis
In a study designed to test the role of extracellular heme in SCD pathogenesis , Ghosh et al. made a serendipitous observation that provides insight into how the concentration of heme is elevated in SCD plasma: heme is amplified. The investigators infused groups of SS, AS, and AA mice with a bolus of purified heme, and as expected this raised the TPH in all the animals to a similar concentration immediately after the challenge. Paradoxically, the TPH in the SS mice continued to increase surpassing the bolus by ~50 % within 30 min. Meanwhile, both control animals (AS and AA) rapidly cleared the exogenous heme from their circulation. This finding showed that there is auto-amplification of extracellular heme in SCD plasma . More importantly, it uncovered for the first time a phenomenon of extracellular heme crisis; an acute uncontrolled release of heme into the circulation that was associated with: (a) enhanced oxidation of intracellular HbS, (b) impaired reduction of intracellular metHbS, (c) enhanced heme-induced hemolysis, and (d) rapid depletion of hemopexin. This phenomenon promoted the development of a lethal ALI reminiscent of ACS in the SS mice [54]. Extracellular heme crisis may be inevitable specifically within occluded vascular segments with impaired blood flow in patients with severe VOC who develop acute hemolysis.
Neutrophil Extracellular Traps (NETs)
NETs are released into the plasma by activated neutrophils. They consist of a mesh of extracellular DNA, nucleosomes, and histones decorated with granular enzymes, typically neutrophil elastase. It is thought that NETs trap pathogens with their high concentrations of enzymes . They were first implicated in SCD pathogenesis in a prospective study including 70 patients with VOC [84]. During VOC, levels of both nucleosomes and elastase-α1-antitrypsin complexes increased significantly as compared to steady-state values. Levels of nucleosomes correlated significantly with elastase-α1-antitrypsin complex levels during painful crisis, and the levels were highest in patients with ACS [84]. Extracellular heme stimulates the release of NETs by increasing intracellular neutrophil ROS formation (Fig. 3.1). Plasma from both humans and mice with SCD promotes NET release in vitro, and infusions of hemopexin reduced the number of pulmonary NETs, and the concentration of plasma nucleosomes in the Berkeley sickle mouse. The production of NETs by neutrophils during TNF-α exposure led to an ACS-like condition and sudden death in these animals following cremasteric surgical preparation [78]. DNase I treatment prolonged survival however hemopexin infusions did not, probably, because of the relatively low dose that was used in this study (0.15 mg hemopexin per mouse) [78]. In SCD patients, infections, which increase systemic TNF-α, are important contributing factors to ACS development , thus enhancing the idea that NETs may play an important role in the pathobiology of this lung condition.
Preclinical Murine ACS Model
Complete saturation of hemopexin yielding at least ~1.5 μM of extracellular heme that is seemingly free from other plasma binding partners triggers a lethal ALI in mice [54]. Given the low plasma hemopexin reserve in SCD, this ALI is inducible in ~70 % of sickle mice (Townes and Berkeley) with a relatively low dose of purified heme (i.v. 35 μmol/kg). This preclinical ACS model shares several similarities with the condition in humans. It is characterized by acute onset, acute anemia, pulmonary infiltration, hypoxemia, hypercapnia, acidosis, pulmonary edema, and sudden death (Table 3.1). This model suggests that continuous supply of extracellular heme fuels ACS, and that targeting this supply can avert respiratory failure even when the initial insult has caused some lung injury. Indeed, this may be one mechanism through which blood transfusion halts ACS, by reducing the concentration of extracellular heme in the intravascular space below the threshold required to cause endothelial barrier disruption.
Table 3.1
Characteristics of ACS in patients and in a preclinical mouse model
Parameter | Patients | Mice |
|---|---|---|
Etiology/trigger | Multiple | Extracellular heme |
Presentation | Acute onset | Acute onset |
Mean hemoglobin loss | 0.7–3.6 g/dl | 1.4 g/dl |
Inflammation | New chest infiltrate | Gross pulmonary infiltrates |
Histology | Edema, alveolar wall necrosis, fat emboli | Edema, alveolar wall thickening, hemorrhage |
Physiological dysfunction | Hypoxemia, hypercapnia | Hypoxemia, hypercapnia, acidosis |
Death due to respiratory failure | Up to 9 % (treated) | 0–20 % (treated)a 70 % (untreated)b |
Plasma risk factors | Elevated free heme | Low hemopexin |
Genetic modifiers | HMOX1, COMMD7 | TLR4 |
Targeted therapy | None | Hemopexin, TAK-242 |
Directly targeting heme with recombinant hemopexin infusions (1 mg per mouse) produces almost immediate improvement in oxygen saturation, followed steadily by restoration of breath rate in SS mice with hemolytic crisis and respiratory distress [54]. Hemopexin has as unique ability to rapidly squelch the redox activities of heme [85]. This suggests that reactive oxygen species (ROS) signaling contributes to the acute injury in the murine ACS lung, as has previously been suggested for the human ACS lung [68]. In addition, the lethal lung injury in the murine model requires a functional TLR4, which is redox-sensitive. The TLR4 antagonist TAK-242 is effective in preventing lung injury in the murine ACS model [54].
TLR4 mutant mice are protected from respiratory disease when challenged with a high dose of heme that causes a lethal ALI in congenic control mice. This means that the basic mechanism of the ALI, and perhaps other injuries caused by extracellular heme in SCD is mediated by TLR4. Using bone marrow chimeric mice, TLR4 expressed by nonhematopoietic vascular tissues (most likely the endothelium), has been found to mediate heme-induced ALI/ACS [54], and stasis in sickle mice. The receptor had been a major therapeutic target in sepsis resulting in the development of TAK-242 [86, 87] and Eritoran [88]. Both drugs have failed phase III sepsis trials nonetheless they may be ideal candidates for repurposing in SCD [89–91]. In preclinical studies, TAK-242 was effective in preventing respiratory failure in sickle mice only when it was given before or immediately after the induction of hemolytic crisis [54]. Since a majority of ACS patients present initially with well-defined antecedent acute events that are interceded by hemolysis, acute prophylaxis using TLR4 blockade may be an effective strategy to alter the clinical course of this condition.
Conclusion: A Mechanistic Model of ACS Pathogenesis
A mechanism explaining how ACS develops in SCD is proposed (Fig. 3.2). A large proportion of sickle erythrocytes trapped in occlusions with reduced flow, and shielded from the paltry reserve of plasma scavenger molecules is destroyed during acute exacerbations in SCD. In this setting, the acute hemolysis inevitably yields extracellular heme. This heme species can be amplified in vivo generating an excess pool sufficient to trigger and sustain a robust TLR4 mediated inflammation that ultimately causes a lethal ALI. This ACS model is characterized by a diversity of features; clinical, biological, physiological, and pathological, that is common or complementary in humans and mice with SCD (Table 3.1). It is versatile, reproducible, and readily amenable to multiple drug discovery platforms. Studies expanding its conceptual framework may help to further improve our understanding of the pathogenesis of ACS, and to develop effective targeted therapies based on clearly defined mechanism for this devastating lung condition.
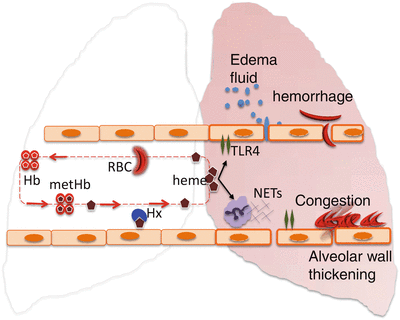
Fig. 3.2
A model of heme-induced acute chest syndrome. The concentration of heme can be auto-amplified in vivo via a phenomenon named extracellular heme crisis (EHC) involving acute hemolysis, release of cell-free hemoglobin, and increased methemoglobin S formation, which ultimately depletes plasma hemopexin reserve. Free extracellular heme interacts with TLR4 on the vessel wall, disrupts the alveolar capillary barrier resulting in acute pulmonary edema, alveolar wall thickening, and microvascular congestion. Collectively these pathologies impair alveolar gas exchange and cause potentially terminal respiratory failure
Acknowledgements
The authors are supported by research funding from the National Heart, Lung and Blood Institute (Award Number: HL106192, HL106192S1, and U01HL117721). The authors thank Sonia Mckoy for comments and proofreading that helped to improve this book chapter.
Authorship
Contribution: OA, AOA, ADA, SG, and SFOA prepared, wrote, and edited the chapter.
Conflict of Interest
The authors OA, AOA, ADA, SG, and SFOA declare that no conflict of interest exists.
References
1.
Charache S, Scott JC, Charache P. “Acute chest syndrome” in adults with sickle cell anemia. Microbiology, treatment, and prevention. Arch Intern Med. 1979;139(1):67–9.PubMed
2.
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–44.PubMed
3.
Gaston M, Rosse WF. The cooperative study of sickle cell disease: review of study design and objectives. Am J Pediatr Hematol Oncol. 1982;4(2):197–201.PubMed
4.
Gaston M, Smith J, Gallagher D, Flournoy-Gill Z, West S, Bellevue R, et al. Recruitment in the cooperative study of sickle cell disease (CSSCD). Control Clin Trials. 1987;8 Suppl 4:131S–40.PubMed
5.
Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette P, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84(2):643–9.PubMed
6.
Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89(5):1787–92.PubMed
7.
Styles LA, Schalkwijk CG, Aarsman AJ, Vichinsky EP, Lubin BH, Kuypers FA. Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood. 1996;87(6):2573–8.PubMed
8.
Styles LA, Aarsman AJ, Vichinsky EP, Kuypers FA. Secretory phospholipase A(2) predicts impending acute chest syndrome in sickle cell disease. Blood. 2000;96(9):3276–8.PubMed
9.
Bargoma EM, Mitsuyoshi JK, Larkin SK, Styles LA, Kuypers FA, Test ST. Serum C-reactive protein parallels secretory phospholipase A2 in sickle cell disease patients with vasoocclusive crisis or acute chest syndrome. Blood. 2005;105(8):3384–5.PubMed
10.
Paul RN, Castro OL, Aggarwal A, Oneal PA. Acute chest syndrome: sickle cell disease. Eur J Haematol. 2011;87(3):191–207.PubMed
11.
Miller ST. How I, treat acute chest syndrome in children with sickle cell disease. Blood. 2011;117(20):5297–305.PubMed
12.
Desai PC, Ataga KI. The acute chest syndrome of sickle cell disease. Expert Opin Pharmacother. 2013;14(8):991–9.PubMed
Related posts:
 Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation
Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation
 Acute Pulmonary Manifestations of Hematologic Malignancies
Acute Pulmonary Manifestations of Hematologic Malignancies
 Leukocyte Kinetics and Migration in the Lungs
Leukocyte Kinetics and Migration in the Lungs
 Storage Lesion: Evolving Concepts and Controversies
Storage Lesion: Evolving Concepts and Controversies
 Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes
Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes
 Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery
Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


