Shiga toxin associated hemolytic uremic syndrome (Stx HUS), a thrombotic microangiopathy, is the most common cause of pediatric acute kidney injury but has no direct treatment. A better understanding of disease pathogenesis may help identify new therapeutic targets. For this reason, the role of complement is being actively studied while eculizumab, the C5 monoclonal antibody, is being used to treat Stx HUS but with conflicting results. A randomized controlled trial would help properly evaluate its use in Stx HUS while more research is required to fully evaluate the role complement plays in the disease pathogenesis.
Key points
- •
Ninety percent of hemolytic uremic syndrome (HUS) occurs after a gastrointestinal infection with a Shiga toxin (Stx)-producing bacterium, usually Escherichia coli O157.
- •
Stx can directly activate the alternative complement pathway and reduce regulatory protein function.
- •
Genetic blockade of the alternative pathway is protective in mice, but therapeutic complement blockade in patients has not been consistently beneficial.
- •
Earlier administration of complement modulatory therapy may be beneficial but a clinical trial is needed to test this hypothesis.
- •
Further work is needed to define the role of complement in the pathogenesis of Stx HUS and direct therapeutic interventions.
Introduction
Hemolytic uremic syndrome (HUS) is characterized by the clinical triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute renal injury. It is the leading single cause of pediatric acute kidney injury. HUS is a type of thrombotic microangiopathy (TMA). Typically, it occurs after a gastrointestinal infection with a Shiga toxin (Stx)-producing pathogen. This diarrhea- or Stx-associated HUS (D+HUS or Stx-HUS) accounts for 90% of cases. Currently, there are no direct treatments and limited prevention strategies. Most patients recover from the acute illness, but there is a 1% to 4% mortality and one-third of patients are left with long-term medical problems. The major challenge facing clinicians and researchers is better understanding the disease pathogenesis in an attempt to develop new therapeutic strategies. With this in mind, attention has turned to advances in the understanding of a related condition, complement-mediated HUS. This atypical HUS subtype is now successfully treated with the C5 monoclonal antibody, eculizumab. Its success prompted clinicians to ask if it could also be used to treat Stx HUS and for researchers to examine the role of complement in the pathogenesis of this disease.
Introduction
Hemolytic uremic syndrome (HUS) is characterized by the clinical triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute renal injury. It is the leading single cause of pediatric acute kidney injury. HUS is a type of thrombotic microangiopathy (TMA). Typically, it occurs after a gastrointestinal infection with a Shiga toxin (Stx)-producing pathogen. This diarrhea- or Stx-associated HUS (D+HUS or Stx-HUS) accounts for 90% of cases. Currently, there are no direct treatments and limited prevention strategies. Most patients recover from the acute illness, but there is a 1% to 4% mortality and one-third of patients are left with long-term medical problems. The major challenge facing clinicians and researchers is better understanding the disease pathogenesis in an attempt to develop new therapeutic strategies. With this in mind, attention has turned to advances in the understanding of a related condition, complement-mediated HUS. This atypical HUS subtype is now successfully treated with the C5 monoclonal antibody, eculizumab. Its success prompted clinicians to ask if it could also be used to treat Stx HUS and for researchers to examine the role of complement in the pathogenesis of this disease.
Epidemiology
Worldwide, Stx-producing Escherichia coli causes 2.8 million illnesses annually. In the United States, it causes more than 265,000 illnesses per year with 3600 hospitalizations and 30 deaths. The global incidence of Stx HUS is 0.2 to 4.28 people per 100,000 population. Seasonal variation occurs with more cases in summer. Stx HUS is more common in children, particularly those under the age of 5. E coli O157, the most common Stx-producing E coli , is responsible for 724 hospitalizations and 11 deaths per year in this age group in the United States.
Clinical course
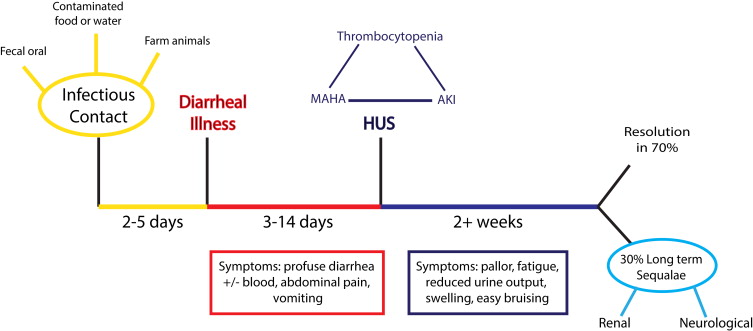
Patients infected with a Stx-producing pathogen usually develop a profuse diarrheal illness 2 to 5 days later ( Fig. 1 ). Signs and symptoms of HUS occur after the diarrhea, but only in 10% to 15% of people infected with E coli O157. The risk of developing Stx HUS is influenced by the organisms serotype, as illustrated in the E coli O104 outbreak, where 22% of infected patients developed HUS. Host factors also play a role ( Box 1 ).
Host factors that increase risk of HUS
Extremes of age (<5 y, >75 y )
Vomiting
>3 days of diarrhea
Blood in stool
High white cell count (>13,000/μL)
Signs and Symptoms
Symptoms of HUS can be vague (see Fig. 1 ). Medical examination reveals signs of oligoanuric kidney injury with fluid overload and hypertension. There is a disease spectrum. More severely affected patients may show signs of other organ involvement, particularly cerebral, pancreatic, hepatic, and cardiac.
Pathology
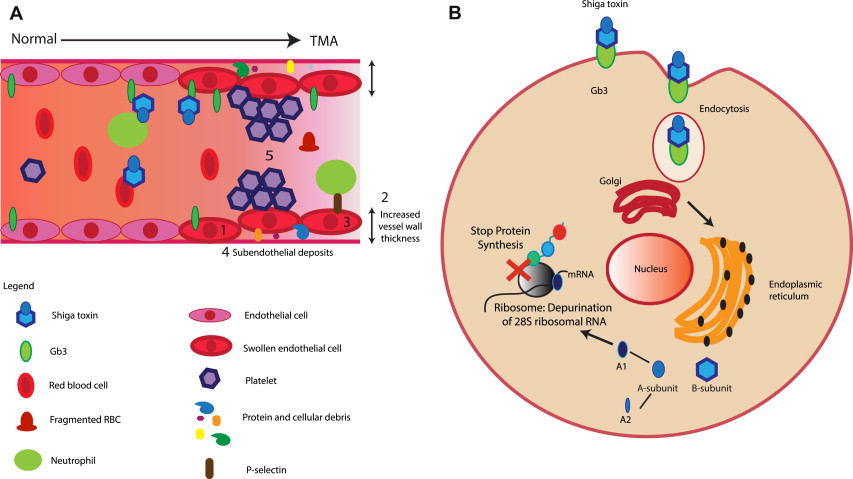
The pathologic feature of HUS is TMA. This term describes characteristic endothelial cell damage ( Fig. 2 A), which leads to complete or partial vessel obstruction, resulting in fragmented red blood cells. Characteristic laboratory findings include consumptive thrombocytopenia and microangiopathic hemolytic anemia. Organ ischemia occurs with symptoms determined by the vascular bed affected. In glomerular TMA, there will be evidence of renal impairment, but if TMA affects the brain neurologic symptoms develop.
Pathogenesis
Stx-producing bacteria infect the large intestine causing colitis. The toxin produced crosses the gastrointestinal epithelium and enters the circulation possibly via globotetraosylceramide (Gb4) cellular receptors, but monocytes and neutrophils may also be involved. Stx targets cells that express globotriosylceramide (Gb3) receptors. Free Stx has never been detected in patient blood. Erythrocytes, neutrophils, and platelets have been implicated in Stx carriage, but this may not be via Gb3 receptors. Recently, it was reported that neutrophils bind Stx via toll-like receptor 4 (TLR4). It is hypothesized that TLR4 has a lower affinity for Stx than Gb3, so when Stx arrives at a Gb3 expressing organ, such as the kidney, it preferentially detaches from the circulating cells and binds to the Gb3-expressing cells.
Cellular Effects
When Stx binds cellular Gb3, the toxin is internalized by endocytosis and, if expressed in a detergent-resistant membrane, undergoes retrograde transport to the endoplasmic reticulum (see Fig. 2 B). Ultimately, Stx stops cellular protein synthesis (see Fig. 2 B) and causes apoptosis with damage to the integrity of the endothelium and exposure of the subendothelial layer leading to TMA. Stx may have other effects, including increasing synthesis of pro-inflammatory cytokines, directly damaging DNA, generating microparticles, causing impaired cellular oxidative balance, and interacting with complement proteins.
The complement system in Shiga toxin-associated hemolytic uremic syndrome pathogenesis
Clinical Evidence of Complement Activation in Shiga Toxin-Associated Hemolytic Uremic Syndrome
Complement activation occurs in Stx HUS patients ( Box 2 ). It may be an early disease feature because activated factor B (Bb) fragments and soluble membrane attack complex (MAC) were detected in patient plasma at the time of hospital admission before starting treatment. One month later, levels had normalized. Platelet-leukocyte complexes and microparticles with evidence of complement activation on their surface have also been identified. These clinical observations show that complement is activated in Stx HUS but not how it contributes to disease pathogenesis.
Low serum C3
C3 breakdown fragments (C3b, C3c, and C3d)
Factor B breakdown fragments (Bb)
Serum-soluble MAC
C3 deposits in kidney
C3/C9 covered platelet-leukocyte complexes and microparticles
In Vitro Evidence
Shiga toxin inhibits protective complement regulators
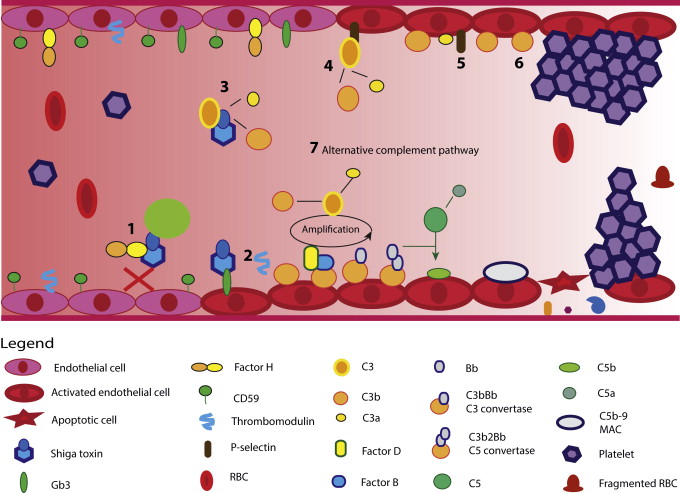
Stx can bind to and inhibit the complement regulatory function of complement factor H (CFH) and CFH-related protein 1, which may make cells vulnerable to complement-mediated damage ( Fig. 3 ). Stx targets the membrane-binding portions of CFH, so although it can still regulate complement in fluid phase, it cannot on the cell surface. This phenomena is the same as that produced by the CFH loss-of-function mutations associated with atypical, complement-mediated HUS. Furthermore, glomerular endothelial cell expression of the membrane-bound complement regulator CD59 and the complement and coagulation regulator thrombomodulin (TM) were reduced by Stx. TM loss may be due to increased cellular shedding, which could explain the high serum TM levels found in Stx HUS patients.
Shiga toxin activates complement
Stx can directly activate complement predominately via the alternative pathway (see Fig. 3 ). Endothelial cells pretreated with Stx and exposed to human serum expressed P-selectin can bind and activate C3 (see Fig. 3 ). The formation of C3a further enhances P-selectin expression, decreases TM expression, and thrombosis ensues. In vitro, C3 complement deposits occurred before thrombin deposition, suggesting that complement deposition could trigger thrombosis. Inhibiting complement activation abolished these effects.
Mouse models
Murine models of Stx HUS are used to study the disease but do not precisely replicate human pathology because mice lack glomerular Gb3 receptors. Instead, they develop renal tubular damage. Heterozygous CFH-deficient mice and C5-deficient mice developed the same tubular damage as wild-type mice, suggesting complement does not play a role in this murine model. Mice treated with higher doses of Stx and lipopolysaccharide (LPS) do develop some intraglomerular platelet clumps and endothelial swelling; this is the best published Stx HUS model to date. In this model, mice have reduced glomerular TM expression with C3 and C9 glomerular deposits, suggesting complement activation. Mice lacking the TM complement regulatory (lectin) domain had a more severe clinical phenotype when challenged with Stx and LPS. Similarly challenged factor B knockout mice showed no C3 deposits and less thrombocytopenia, and renal function was maintained. In this murine model of Stx HUS, alternative pathway blockade through genetic knockout was protective. Further studies suggested a role for complement activation specifically in the specialized glomerular epithelial cell, the podocyte, highlighting that Stx has effects on other cell types too. Further work is needed to confirm these findings in patients.
Related posts:
 Complement in Health and Disease
Complement in Health and Disease
 Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
 Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


